

CO2 Reduction to CO with 19% Efficiency in a Solar-Driven Gas Diffusion Electrode Flow Cell under Outdoor Solar Illumination
- Wen-Hui ChengWen-Hui ChengDepartment of Applied Physics and Material Science, California Institute of Technology, Pasadena, California 91125, United StatesMore by Wen-Hui Cheng,
- Matthias H. RichterMatthias H. RichterDivision of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, California 91125, United StatesMore by Matthias H. Richter,
- Ian SullivanIan SullivanDivision of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, California 91125, United StatesMore by Ian Sullivan,
- David M. LarsonDavid M. LarsonChemical Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, California 94720, United StatesMore by David M. Larson,
- Chengxiang XiangChengxiang XiangDivision of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, California 91125, United StatesMore by Chengxiang Xiang,
- Bruce S. Brunschwig*Bruce S. Brunschwig*Email: [email protected]Beckman Institute, California Institute of Technology, Pasadena, California 91125, United StatesMore by Bruce S. Brunschwig, and
- Harry A. Atwater*Harry A. Atwater*Email: [email protected]Department of Applied Physics and Material Science, California Institute of Technology, Pasadena, California 91125, United StatesMore by Harry A. Atwater


Abstract

{kind=link}
Solar-driven reduction of carbon dioxide represents a carbon-neutral pathway for the synthesis of fuels and chemicals. We report here results for solar-driven CO2 reduction using a gas diffusion electrode (GDE) directly powered by a photovoltaic cell. A GaInP/GaInAs/Ge triple-junction photovoltaic cell was used to power a reverse-assembled gas diffusion electrode employing a Ag nanoparticle catalyst layer. The device had a solar-to-CO energy conversion efficiency of 19.1% under simulated AM 1.5G illumination at 1 Sun. The use of a reverse-assembled GDE prevented transition from a wetted to a flooded catalyst bed and allowed the device to operate stably for >150 h with no loss in efficiency. Outdoor measurements were performed under ambient solar illumination in Pasadena, California, resulting in a peak solar-to-CO efficiency of 18.7% with a CO production rate of 47 mg·cm–2 per day and a diurnal-averaged solar-to-fuel conversion efficiency of 5.8%.
Solar photovoltaic and wind energy conversion are rapidly growing sources of low-carbon electric power.(1) However, the intermittency of solar and wind resources over wide time scales ranging from minutes to months means solar electricity is not a dispatchable power source. Thus, efficient and inexpensive approaches for energy storage are needed for wide penetration of renewable energy into the power grid.(2,3) While electrical energy storage in batteries may be important for short-term storage and grid power management, seasonal energy storage is unlikely to rely on batteries. Transformation of solar energy into chemical bonds provides a long-term energy storage strategy that opens a path for the synthesis of fuels and chemicals.(4) One approach to chemical energy storage is via solar-driven hydrogen generation, where (i) photovoltaics supply carbon-free electricity to the grid that is used to generate H2 by water electrolysis at high current densities;(5) (ii) photovoltaics are used to directly drive electrolysis at low current densities,(6) or (iii) an integrated photoelectrochemical device performs unassisted direct water splitting to form H2.(7,8) Parallel to solar hydrogen generation approaches, pathways for solar-driven reduction of carbon dioxide to fuels have used (i) direct electrolysis,(9) (ii) photovoltaic directly driven electrolysis,(10) and (iii) integrated photoelectrochemical conversion.(11,12) Of particular interest is solar-driven reduction of carbon dioxide using a high-efficiency photovoltaic (PV) device directly coupled to an electrochemical cell tailored for reduction of CO2 to CO.(13,14) Mixtures of solar-generated CO and H2(15) could be used as syngas precursors in a future Fischer–Tropsch chemical synthesis process(16) to produce high molecular weight hydrocarbon fuels or chemicals as products.(17) Carbon dioxide reduction to CO is generally more energy efficient and kinetically easier than direct reduction of CO2 to multicarbon products.(14,18,19)
Among the most efficient heterogeneous solid-state catalysts for CO2 reduction to CO are gold,(20,21) silver,(22) WSe2,(23) and MoS2.(24) The use of high surface area morphology structures such as nanoparticles can improve catalytic activity.(25) Other factors that impact catalytic performance include catalyst morphology,(20) cations present in the electrolyte solution,(26) electrolyte concentration,(27) and local pH.(28) The state-of-the-art CO2-to-CO conversion using a Au needle catalyst(27) showed an operating current of 15 mA·cm–2 and 95% Faradaic efficiency at −0.35 V vs RHE. However, the current record efficiency device for solar conversion of CO2 to CO using a solution-based electrochemical cell suffered from low current density (0.33 mA·cm–2 at −0.6 V vs RHE) due to limited catalyst activity. This required the use of large-area electrodes to match the photovoltaic device area.(10)Table S1 shows overpotential and Faradaic efficiency data at current densities close to 15 mA·cm–2 along with the electrolyte conditions and catalyst loading for various Ag and Au electrodes. The catalytic activities shown in Table S1 indicate that in many cases nanoparticles of Ag have an activity similar to that of Au while costing significantly less.
Bulk aqueous electrolyte cells can exhibit high catalyst overpotentials due to the limited solubility of CO2 (33.4 mM) in the electrolyte, a limited pH operating range of ∼6–10, and slow ionic transport in the solution. In contrast, gas diffusion electrode (GDE) assemblies do not suffer these same restrictions.(29−35) In a GDE using 1 atm CO2 vapor, CO2 is transported in the vapor phase and reacts at a thin (<100 nm) solid–liquid–gas phase interface. In this configuration, liquid-state concentration and diffusion do not limit the conversion rate, resulting in lower overpotentials and higher current densities for CO2 reduction.(30) Simulations have also shown that a cell using a thin (10 nm) layer of electrolyte on the catalysts (wetted catalyst) outperforms cells with either a completely dry or a completely flooded catalyst configuration.(36) These insights have led to the development of gas diffusion electrodes(37) and membrane electrode assemblies (MEA)(38) with a humidified gas supply to facilitate ion conduction and water balance.
Although membrane electrode assembly systems are more scalable, they often suffer from short-term stability due to salt precipitation or membrane dehydration at high current densities.(39) Hence, we chose to work with an aqueous GDE cell configuration. In this work, we employ a triple-junction photovoltaic (PV) device directly coupled with a gas diffusion electrode (GDE) as the first demonstration of an electrolyte flow type PV-GDE reactor that provides both high selectivity and long-term stability. For a directly driven PV-GDE system, the power generated by the PV is directly supplied to the GDE. In our device, the areas of the PV photoabsorber (APV) and GDE (AGDE) were both 0.31 cm2. To match the lower current density of the PV cell with the operating conditions of the anode, a relatively low catalyst loading of GDE was chosen. A Ag nanoparticle catalyst was used because of its relatively high activity and relatively low cost (Table S1).
Figure 1a is an illustration of the compression flow cell employed for the evaluation of gas diffusion electrode catalytic performance. Dilute silver nanoparticles (Ag-NPs) with diameters of ≤50 nm were drop cast onto the microporous side of the GDE substrate (Sigracet 29BC). The loading of Ag-NPs in this work was measured to be 0.12 mg·cm–2. A detailed description can be found in Methods in the Supporting Information. Scanning electron microscopy (SEM) images of the microporous layer with and without Ag-NPs are shown in Figure 1b. Gas was delivered to the GDE through an interdigitated electrode flow field (Figures 1a and S1) against which the GDE is compressed to maximize the interaction of CO2 with the catalyst and gas utilization.(40) Current to the GDE was supplied through the interdigitated electrode to Ag-NP/carbon paper substrate. Gaseous products were collected at the outlet of the flow field, which was directly connected to a gas chromatograph (for more information see Methods in the Supporting Information).
Figure 1

Figure 1. Gas diffusion electrode with Ag-NP catalyst. (a) Cell configuration composed of (1) NiOx or Pt anode, (2) Ag-NPs on Sigracet 29BC carbon paper cathode, (3) anion exchange membrane, (4) CO2 gas inlet and CO/CO2 outlet, (5) acrylic backplate, (6) catholyte chamber, (7) anolyte chamber, (8) reference electrode, (9) GDE (cathode) power connector, and (10) anode power connector. Black arrows indicate the gas flow, and white arrows indicate the electrolyte flow. Note that the backplate (5) is designed to use an interdigitated wire electrode flow field to enhance the interaction between gas and catalysts and improve CO2 utilization (see also Figure S1). (b) Scanning electron microscopy images of carbon paper without (top) and with (bottom) Ag-NP catalyst, secondary electrons image (left row) backscattered electrons image (right row). (c) Illustration of the reverse-assembled GDE cathode cross-section with wetted catalyst and operation for CO2 reduction.
{kind=link}
An issue for aqueous GDEs is flooding or saturation of the porous catalyst layer with electrolyte or water during operation. This results in a thick (>1 μm) electrolyte layer and a diffusion-limited supply of CO2 to the electrode.(41) To maintain the catalyst in a wetted but not flooded condition that minimizes losses of CO2 to the electrolyte and extends the operational lifetime, we assembled our aqueous GDE in a nontraditional manner with the catalyst coating of Ag-NPs facing away from the electrolyte and toward the CO2 gas supply. We denote this configuration as a reverse-assembled GDE. The microporous layer of the GDE was treated with polytetrafluoroethylene (PTFE), which helped to prevent flooding. Needle valves in the gas and liquid output streams allowed separation of the liquid and gas phases as well as control of the pressure difference between the aqueous electrolyte and the CO2 stream. Contact angle analysis indicated that the Ag-NP coated surface was significantly less hydrophobic than the surface without Ag-NPs. Contact angle and optical microscope images of the GDE are shown in Figure S2.
With both the gas inlet and outlet on the same side of the GDE, the device operates in a “flow-by” GDE configuration. The Ag-NP catalyst side of the electrode was facing the CO2 gas channel as illustrated in Figure 1c. This orientation of the Ag-NPs maintained a thin electrolyte layer on the catalyst and enhanced the rate of CO2 reduction.(36) The turnover frequency of the Ag-NP catalyst for the reverse-assembled GDE at −0.6 V vs RHE was calculated as ∼9 × 103 h–1 (see the Supporting Information). The anode was made from either Pt or an electrochemically activated Ni foam for three- and two-electrode measurements, respectively. An aqueous catholyte of 1 M aqueous potassium bicarbonate (KHCO3) or potassium hydroxide (KOH) was used under near neutral or basic conditions, respectively. In all cases, 1 M KOH was the anolyte. The anion exchange membrane (AEM) was Selemion for neutral environment or Fumasep FAA-3-50 for alkaline environment. Electrolyte (500 mL) was continuously pumped through the cathode chamber in a closed loop at a rate of 2 mL·min−1. A change of pH (from 14 to 13.7) was observed for the 1 M KOH catholyte after 150 h of continuous operation, corresponding to irreversible loss of 0.25 mol KOH (50% of the electrolyte; see the Supporting Information). Further improvement to reduce CO2 loss or regenerate the electrolyte would be necessary for fully sustainable operation. The neutralized carbonate electrolyte can possibly be utilized in a carbonate-to-syngas system to compensate the loss of CO2 in a gas-fed MEA cell with a bipolar membrane.(42)
Results from three-electrode measurements for reverse- and standard-assembled GDEs are shown in panels a and b of Figure 2, respectively, for 1 M KHCO3 (bulk pH of 8.5) and 1 M KOH (bulk pH of 14). Current densities are substantially lower than for earlier reported GDE devices because of the low catalyst loading used to match the current from the PV (current matching). For the reverse-assembled GDE, both the Faradaic efficiency (fFE,CO) for CO and current density (JGDE) increased with increasing potential with fFE,CO close to 100% at −0.6 V vs RHE in 1 M KOH (Figure 2a). Similar trends of current density and Faradaic efficiency versus applied potential were found for the standard-assembled GDE (Figure 2b). To compare the activity of the Ag-NPs in different orientations and pH, overpotential analysis for CO2 reduction to CO was preformed (Figure 2c). The comparable Tafel slopes (∼0.23 V/dec) in KHCO3 and KOH for either orientation indicate a similar catalytic pathway regardless of the operating conditions. The Tafel behavior plotted with potentials vs NHE falls on a rough single line (Figure S3) and suggests that the rate-determining step for the reduction on our Ag-NP GDE is not proton-limited. The achievable current density and Faradaic efficiency (fFE,CO) for CO are higher in 1 M KOH than in 1 M KHCO3 at the same overpotential (Figure 2c), likely because of a pH-independent rate-determining step. All subsequent measurements were, therefore, performed using 1 M KOH for the PV-GDE integrated device.
Figure 2

Figure 2. Dark catalysis three-electrode measurement of Ag-NPs GDE. Faradaic efficiency versus GDE potential operated in 1 M KHCO3 (left half of graph) or 1 M KOH (right half of graph) of (a) the reserve-assembled Ag-NP GDE and (b) a standard-assembled Ag-NP GDE. (c) Overpotential versus CO partial current of Ag-NPs GDE for CO2 reduction to CO. Overpotential = |UGDE,RHE + 0.11 V|, JCO ≡ JGDE × fFE,CO. (d) Stability of reserve-assembled and standard-assembled Ag-NPs GDE operated at −0.6 V vs RHE in 1 M KOH.
{kind=link}
Figure 2d shows the Faradaic efficiency for CO versus time at −0.6 V vs RHE for the two GDE orientations in KOH. For the standard configuration, the fFE,CO decreased to ∼75% after 1 h and to 50% after 2 h, while for the reverse configuration, the fFE,CO was ∼97% for 3 h. Though similar in initial current density and fFE,CO, the standard assembly, with the Ag-NP catalyst facing the electrolyte, became flooded during the first hour of operation resulting in a reduction of the Faradaic efficiency.
We performed two-electrode measurements for the GDE using an electrochemically activated nickel foam anode coupled to the GaInP/GaInAs/Ge triple-junction cell. For detailed information about the solar cell see Methods Figures S4 and S5, and Table S2 in the Supporting Information. A schematic of the cell is shown in Figure 3a with 1 M KOH as electrolyte using a Fumasep FAA-3-50 membrane. Both the cell potential (Ucell) and the cathode-to-reference electrode potential (UGDE) were monitored during the operation. We calculated the solar-to-fuel efficiency (ηSTF) for CO2 reduction using eq 1. (1)where ΔUrxn is the thermodynamic potential difference between the oxygen evolution half reaction (OER) and the CO2 reduction half reaction of 1.34 V, A the area of the GDE or PV with AGDE = APV = 0.31 cm2, J (= JGDE = JPV) the operation current density of the system, and Plight the incident light irradiance (mW·cm–2) on the photovoltaic cell. The energy efficiency for the GDE cell (ηGDE) was defined as follows:
(1)where ΔUrxn is the thermodynamic potential difference between the oxygen evolution half reaction (OER) and the CO2 reduction half reaction of 1.34 V, A the area of the GDE or PV with AGDE = APV = 0.31 cm2, J (= JGDE = JPV) the operation current density of the system, and Plight the incident light irradiance (mW·cm–2) on the photovoltaic cell. The energy efficiency for the GDE cell (ηGDE) was defined as follows: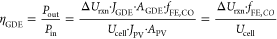 (2)where JGDE·AGDE = JPV·APV and Ucell is the total operating voltage of the cell.
(2)where JGDE·AGDE = JPV·APV and Ucell is the total operating voltage of the cell.
Figure 3

Figure 3. Light driven PV-GDE measurement (APV = AGDE = 0.31 cm2). (a) Illustration of wire connection between the triple-junction cell and GDE cell. (b) J–U characteristic of Ni anode, solar cell with Ni anode, and Ag-NP gas diffusion cathode under 1 Sun. (c) Current, GDE potential vs RHE, and cell voltage measurement over 20 h duration. (d) Corresponding CO Faradaic efficiency and solar-to-fuel efficiency over the same 20 h duration.
{kind=link}
To evaluate the efficiency and stability, we measured cell parameters using simulated AM 1.5G sun illumination at 1 Sun in the laboratory, as shown in Figure 3b–d. The blue curve in Figure 3b represents the performance of the electrochemically activated Ni foam anode alone, while the yellow curve indicates the behavior of PV plus anode. The red curve shows the catalytic current of the Ag-NPs GDE. The intersection between the red and yellow curves in Figure 3b defines the operation point, located at −0.6 V vs RHE and 14.4 mA·cm–2 with a cell voltage of 2.23 V. Panels c and d of Figure 3 illustrate the cell performance over 20 h with an average Faradaic efficiency for CO of 99 ± 2% and an average CO production rate of 2.3 mg·h−1. No degradation in performance was observed. From the experimental results, we calculated the average solar-to-CO efficiency for the 20 h operation as 19.1 ± 0.2%, with an average energy efficiency ηGDE of 59.4 ± 0.6%. The error bars were obtained as the variation within the 20 h of operation. All the experimental results are summarized in Table S3. The chemical composition of the Ag-NP catalyst layer was examined before and after the reaction by X-ray photoelectron spectroscopy as shown in Figure S6. No obvious changes were observed other than the absorption of potassium after operation with the Ag-NP catalyst maintaining its metallic phase.
The solar-to-CO efficiency of 19.1% represents a new record efficiency. A performance comparison with the current state-of-the-art PV-electrolyzer for CO2 reduction to CO is shown in Table S4. The PV-GDE device had a CO production rate per projected cathode area 50 times higher than for the bulk electrolyte device (7.4 mg·h–1·cm–2 versus 0.145 mg·h–1·cm–2) with greatly improved stability (20 h with no degradation versus 15% loss in 5 h).(10) A similar PV-GDE device operated under 3.25 Suns illumination with AGDE = 1 cm2 and APV = 0.31 cm2 (3.25 ≈ AGDE/APV) showed over 150 h of stability, with an average Faradaic efficiency of 96 ± 2%, an average solar-to-CO efficiency of 18.9 ± 0.5%, and an average energy efficiency ηGDE of 53.7 ± 1.2% (Figure S7).
Full day outdoor tests were conducted with online gas product analysis in order to obtain the solar-to-fuel efficiency over the entire day. Results are shown in Figure 4. The triple-junction cell and a calibrated silicon photodiode were mounted on a solar tracker to maintain optimum orientation toward the Sun (see illustration in Figure S8). The dips in sun intensity at 7:00–9:00 a.m. and 4:00–6:00 p.m. in the data were the result of trees blocking the sunlight. The system operated at a cell voltage of 2.20 V and GDE potential of −0.57 V vs RHE under natural full sun illumination. A Faradaic efficiency of 96 ± 8% and solar-to-fuel conversion efficiency of 18.7 ± 1.7% was observed over an optimal 6 h period within the day. The diurnal-averaged solar-to-fuel conversion efficiency was 5.8%. The CO production rate for 1 day under actual outdoor sun conditions was calculated to be 15 mg·day−1 of CO. Another outdoor demonstration used a lens to concentrate the sunlight, producing an irradiance of 3.25 Suns (C = 3.25, AGDE = 1 cm2, APV = 0.31 cm2) with data included in Figure S9 and Table S3 with a CO generation rate of 50 mg·day−1. Using this calculated rate, a system scale up to 1 m2 would result in a CO production rate of 0.5 kg·day−1.
Figure 4

Figure 4. Outdoor assessments of solar-driven PV-GDE in Pasadena, CA (APV = AGDE = 0.31 cm2). The solar irradiance was monitored with a calibrated silicon photodiode. Operating current density J (= JGDE = JPV), cell voltage Ucell, GDE potential UGDE vs RHE, CO Faradaic efficiency fFE,CO, and solar-to-fuel efficiency ηSTF were recorded for a 24 h day cycle.
{kind=link}
The performance of our directly coupled PV-GDE device was compared to a DC–DC converter coupled PV and GDE with power-matching electronics. We simulate DC–DC converter output curves with the input of our solid-state PV curve as shown in Figure S10. Though the DC–DC converter can track the maximum power point (MPP) of the PV, a practical loss of 5–10% is expected.(43) The operating point for the directly driven PV-GDE cell is Ucell = 2.23 V and J = 14.4 mA·cm–2 with a maximum efficiency of 19.3%. With a 95% efficient DC–DC converter, the operation point would be Ucell = 2.22 V and J = 13.8 mA·cm–2 with a maximum efficiency of 18.5%. For a 90% efficient DC–DC converter, the operation point would be Ucell = 2.20 V and J = 13.2 mA·cm–2 with a maximum efficiency of 17.7%. The maximum efficiencies are calculated assuming 100% CO Faradaic efficiency. All systems are summarized in Table S3. The slightly higher efficiency of our directly driven PV-GDE device, compared to the same setup with integrated DC–DC converter and power matching electronics, reveals the potential of developing a directly coupled PV-GDE device with its reduced complexity.
In summary, we have demonstrated a highly efficient solar-driven CO2 reduction device for CO generation using a flow-by reverse-assembled gas diffusion electrode cell directly coupled to a triple-junction solar cell. The reverse-assembled GDE is designed to minimize parasitic CO2 losses, utilizing a high CO2 concentration and low overpotential catalysts for the CO2 reduction reaction. The Ag-NPs-based catalyst exhibited near unity Faradaic efficiency toward CO generation at approximately −0.6 V vs RHE in 1 M KOH electrolyte. The PV-GDE system was evaluated under both laboratory AM 1.5G simulated solar irradiation and outdoor real sun conditions. Near-unity Faradaic efficiency was observed for CO2-to-CO conversion, and an average solar-to-CO energy efficiency of 19.1% was achieved with AM 1.5G illumination at 1 Sun, leading to a CO production rate per catalyst area over 50 times higher than that of the current record photovoltaic-driven electrolysis device. The GDE was demonstrated to be stable for over 150 h without degradation, supporting our hypothesis that, by using a reverse-assembled GDE device configuration with the catalyst layer facing toward the CO2 gas supply, we could extend the system operation time without suffering a transition from a wetted to a flooded gas diffusion layer. Under outdoor sun conditions, the PV-GDE system exhibited a solar-to-CO conversion efficiency of 18.7% during noontime and yielded a CO production rate of 15 mg·cm–2 per day. This reverse-assembled PV-GDE establishes a new efficiency record for directly solar-driven CO2 reduction and offers an example of a very high-efficiency, stable device for solar CO2 conversion.
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsenergylett.9b02576.
Methods, calculations of solar-to-fuel efficiency, GDE efficiency, turnover frequency, cell potentials, CO2 loss, and supporting figures and tables (PDF)
Terms & Conditions
Electronic Supporting Information files are available without a subscription to ACS Web Editions. The American Chemical Society holds a copyright ownership interest in any copyrightable Supporting Information. Files available from the ACS website may be downloaded for personal use only. Users are not otherwise permitted to reproduce, republish, redistribute, or sell any Supporting Information from the ACS website, either in whole or in part, in either machine-readable form or any other form without permission from the American Chemical Society. For permission to reproduce, republish and redistribute this material, requesters must process their own requests via the RightsLink permission system. Information about how to use the RightsLink permission system can be found at http://pubs.acs.org/page/copyright/permissions.html.
Author Information
- Harry A. Atwater - Department of Applied Physics and Material Science, California Institute of Technology, Pasadena, California 91125, United States;
 http://orcid.org/0000-0001-9435-0201;
Email: [email protected]
http://orcid.org/0000-0001-9435-0201;
Email: [email protected]
- Wen-Hui Cheng - Department of Applied Physics and Material Science, California Institute of Technology, Pasadena, California 91125, United States;http://orcid.org/0000-0003-3233-4606
- Matthias H. Richter - Division of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, California 91125, United States;http://orcid.org/0000-0003-0091-2045
- Ian Sullivan - Division of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, California 91125, United States;http://orcid.org/0000-0003-0632-4607
- Chengxiang Xiang - Division of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, California 91125, United States;http://orcid.org/0000-0002-1698-6754
W.-H.C., M.H.R., and H.A.A. conceived of the experimental study. W.-H.C. and M.H.R. executed the experiments and did the data analysis. D.M.L., I.S., B.S.B., and C.X. provided technical support and scientific discussion. W.-H.C., M.H.R., B.S.B., and H.A.A. wrote the Letter, and all authors commented on the manuscript.
The authors declare no competing financial interest.
Acknowledgments
This work was supported through the Office of Science of the U.S. Department of Energy (DOE) under award no. DE SC0004993 to the Joint Center for Artificial Photosynthesis, a DOE Energy Innovation Hub. Research was in part carried out at the Molecular Materials Research Center of the Beckman Institute of the California Institute of Technology.
References
This article references 43 other publications.
- 1Davis, S. J.; Lewis, N. S.; Shaner, M. R.; Aggarwal, S.; Arent, D.; Azevedo, I. L.; Benson, S. M.; Bradley, T.; Brouwer, J.; Chiang, Y.-M.; Clack, C. T. M.; Cohen, A.; Doig, S.; Edmonds, J.; Fennell, P.; Field, C. B.; Hannegan, B.; Hodge, B.-M.; Hoffert, M. I.; Ingersoll, E.; Jaramillo, P.; Lackner, K. S.; Mach, K. J.; Mastrandrea, M.; Ogden, J.; Peterson, P. F.; Sanchez, D. L.; Sperling, D.; Stagner, J.; Trancik, J. E.; Yang, C.-J.; Caldeira, K. Net-Zero Emissions Energy Systems. Science 2018, 360 (6396), eaas9793 DOI: 10.1126/science.aas9793
- 2Armstrong, R. C.; Wolfram, C.; de Jong, K. P.; Gross, R.; Lewis, N. S.; Boardman, B.; Ragauskas, A. J.; Ehrhardt-Martinez, K.; Crabtree, G.; Ramana, M. V. The Frontiers of Energy. Nature Energy 2016, 1 (1), 15020, DOI: 10.1038/nenergy.2015.20
- 3Shaner, M. R.; Davis, S. J.; Lewis, N. S.; Caldeira, K. Geophysical Constraints on the Reliability of Solar and Wind Power in the United States. Energy Environ. Sci. 2018, 11 (4), 914– 925, DOI: 10.1039/C7EE03029K
- 4Lewis, N. S. Toward Cost-Effective Solar Energy Use. Science 2007, 315 (5813), 798– 801, DOI: 10.1126/science.1137014[Crossref], [PubMed], [CAS], Google Scholar4https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BD2sXhsVShsrs%253D&md5=cc048d85ee8244f9d0d58802cc124395Toward Cost-Effective Solar Energy UseLewis, Nathan S.Science (Washington, DC, United States) (2007), 315 (5813), 798-801CODEN: SCIEAS; ISSN:0036-8075. (American Association for the Advancement of Science)A review. At present, solar energy conversion technologies face cost and scalability hurdles in the technologies required for a complete energy system. To provide a truly widespread primary energy source, solar energy must be captured, converted, and stored in a cost-effective fashion. New developments in nanotechnol., biotechnol., and the materials and phys. sciences may enable step-change approaches to cost-effective, globally scalable systems for solar energy use.
- 5Gray, E. M.; Webb, C. J.; Andrews, J.; Shabani, B.; Tsai, P. J.; Chan, S. L. I. Hydrogen Storage for Off-Grid Power Supply. Int. J. Hydrogen Energy 2011, 36 (1), 654– 663, DOI: 10.1016/j.ijhydene.2010.09.051[Crossref], [CAS], Google Scholar5https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC3MXht1Gjt7w%253D&md5=39d2a80bd405461715d573f44da5ea62Hydrogen storage for off-grid power supplyGray, E. MacA.; Webb, C. J.; Andrews, J.; Shabani, B.; Tsai, P. J.; Chan, S. L. I.International Journal of Hydrogen Energy (2011), 36 (1), 654-663CODEN: IJHEDX; ISSN:0360-3199. (Elsevier Ltd.)The use of intermittent renewable energy sources for power supply to off-grid electricity consumers depends on energy storage technol. to guarantee continuous supply. Potential applications of storage-guaranteed systems range from small installations for remote telecoms, water-pumping and single dwellings, to farms and whole communities for whom grid connection is too expensive or otherwise infeasible, to industrial, military and humanitarian uses. In this paper we explore some of the tech. issues surrounding the use of hydrogen storage, in conjunction with a PEM electrolyzer and PEM fuel cell, to guarantee electricity supply when the energy source is intermittent, most typically solar photovoltaic. We advocate metal-hydride storage and compare its energy d. to that of Li-ion battery storage, concluding that a significantly smaller package is possible with metal-hydride storage. A simple approach to match the output of a photovoltaic array to an electrolyzer is presented. The properties required for the metal-hydride storage material to interface the electrolyzer to the fuel cell are discussed in detail. Relatively conventional Mischmetal-based AB5 alloys are suitable for this application.
- 6Jia, J.; Seitz, L. C.; Benck, J. D.; Huo, Y.; Chen, Y.; Ng, J. W. D.; Bilir, T.; Harris, J. S.; Jaramillo, T. F. Solar Water Splitting by Photovoltaic-Electrolysis with a Solar-to-Hydrogen Efficiency Over 30%. Nat. Commun. 2016, 7, 13237, DOI: 10.1038/ncomms13237[Crossref], [PubMed], [CAS], Google Scholar6https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC28XhvVSisLfI&md5=2e1f9254a9aa1862b6a03635a0429949Solar water splitting by photovoltaic-electrolysis with a solar-to-hydrogen efficiency over 30%Jia, Jieyang; Seitz, Linsey C.; Benck, Jesse D.; Huo, Yijie; Chen, Yusi; Ng, Jia Wei Desmond; Bilir, Taner; Harris, James S.; Jaramillo, Thomas F.Nature Communications (2016), 7 (), 13237CODEN: NCAOBW; ISSN:2041-1723. (Nature Publishing Group)Hydrogen prodn. via electrochem. water splitting is a promising approach for storing solar energy. For this technol. to be economically competitive, it is crit. to develop water splitting systems with high solar-to-hydrogen (STH) efficiencies. Here we report a photovoltaic-electrolysis system with the highest STH efficiency for any water splitting technol. to date, to the best of our knowledge. Our system consists of two polymer electrolyte membrane electrolyzers in series with one InGaP/GaAs/GaInNAsSb triple-junction solar cell, which produces a large-enough voltage to drive both electrolyzers with no addnl. energy input. The solar concn. is adjusted such that the max. power point of the photovoltaic is well matched to the operating capacity of the electrolyzers to optimize the system efficiency. The system achieves a 48-h av. STH efficiency of 30%. These results demonstrate the potential of photovoltaic-electrolysis systems for cost-effective solar energy storage.
- 7Young, J. L.; Steiner, M. A.; Döscher, H.; France, R. M.; Turner, J. A.; Deutsch, T. G. Direct Solar-to-Hydrogen Conversion via Inverted Metamorphic Multi-Junction Semiconductor Architectures. Nature Energy 2017, 2, 17028, DOI: 10.1038/nenergy.2017.28[Crossref], [CAS], Google Scholar7https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC2sXot1ykuro%253D&md5=04ac8805e7de2455968709947216ebbcDirect solar-to-hydrogen conversion via inverted metamorphic multi-junction semiconductor architecturesYoung, James L.; Steiner, Myles A.; Doscher, Henning; France, Ryan M.; Turner, John A.; Deutsch, Todd G.Nature Energy (2017), 2 (4), 17028CODEN: NEANFD; ISSN:2058-7546. (Nature Publishing Group)Solar water splitting via multi-junction semiconductor photoelectrochem. cells provides direct conversion of solar energy to stored chem. energy as hydrogen bonds. Economical hydrogen prodn. demands high conversion efficiency to reduce balance-of-systems costs. For sufficient photovoltage, water-splitting efficiency is proportional to the device photocurrent, which can be tuned by judicious selection and integration of optimal semiconductor bandgaps. Here, we demonstrate highly efficient, immersed water-splitting electrodes enabled by inverted metamorphic epitaxy and a transparent graded buffer that allows the bandgap of each junction to be independently varied. Voltage losses at the electrolyte interface are reduced by 0.55 V over traditional, uniformly p-doped photocathodes by using a buried p-n junction. Advanced on-sun benchmarking, spectrally cor. and validated with incident photon-to-current efficiency, yields over 16% solar-to-hydrogen efficiency with GaInP/GaInAs tandem absorbers, representing a 60% improvement over the classical, high-efficiency tandem III-V device.
- 8Cheng, W.-H.; Richter, M. H.; May, M. M.; Ohlmann, J.; Lackner, D.; Dimroth, F.; Hannappel, T.; Atwater, H. A.; Lewerenz, H. J. Monolithic Photoelectrochemical Device for Direct Water Splitting with 19% Efficiency. ACS Energy Lett. 2018, 3 (8), 1795– 1800, DOI: 10.1021/acsenergylett.8b00920[ACS Full Text
 ], [CAS], Google Scholar8https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC1cXhtFyqtbrK&md5=0597725a2cef1271ca423c4764a23c9cMonolithic Photoelectrochemical Device for Direct Water Splitting with 19% EfficiencyCheng, Wen-Hui; Richter, Matthias H.; May, Matthias M.; Ohlmann, Jens; Lackner, David; Dimroth, Frank; Hannappel, Thomas; Atwater, Harry A.; Lewerenz, Hans-JoachimACS Energy Letters (2018), 3 (8), 1795-1800CODEN: AELCCP; ISSN:2380-8195. (American Chemical Society)Efficient unassisted solar water splitting, a pathway to storable renewable energy in the form of chem. bonds, requires optimization of a photoelectrochem. device based on photovoltaic tandem heterojunctions. We report a monolithic photocathode device architecture that exhibits significantly reduced surface reflectivity, minimizing parasitic light absorption and reflection losses. A tailored multifunctional cryst. titania interphase layer acts as a corrosion protection layer, with favorable band alignment between the semiconductor conduction band and the energy level for water redn., facilitating electron transport at the cathode-electrolyte interface. It also provides a favorable substrate for adhesion of high-activity Rh catalyst nanoparticles. Under simulated AM 1.5G irradn., solar-to-H efficiencies of 19.3 and 18.5% are obtained in acidic and neutral electrolytes, resp. The system reaches 0.85 of the theor. limit for photoelectrochem. water splitting for the energy gap combination employed in the tandem-junction photoelectrode structure.
], [CAS], Google Scholar8https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC1cXhtFyqtbrK&md5=0597725a2cef1271ca423c4764a23c9cMonolithic Photoelectrochemical Device for Direct Water Splitting with 19% EfficiencyCheng, Wen-Hui; Richter, Matthias H.; May, Matthias M.; Ohlmann, Jens; Lackner, David; Dimroth, Frank; Hannappel, Thomas; Atwater, Harry A.; Lewerenz, Hans-JoachimACS Energy Letters (2018), 3 (8), 1795-1800CODEN: AELCCP; ISSN:2380-8195. (American Chemical Society)Efficient unassisted solar water splitting, a pathway to storable renewable energy in the form of chem. bonds, requires optimization of a photoelectrochem. device based on photovoltaic tandem heterojunctions. We report a monolithic photocathode device architecture that exhibits significantly reduced surface reflectivity, minimizing parasitic light absorption and reflection losses. A tailored multifunctional cryst. titania interphase layer acts as a corrosion protection layer, with favorable band alignment between the semiconductor conduction band and the energy level for water redn., facilitating electron transport at the cathode-electrolyte interface. It also provides a favorable substrate for adhesion of high-activity Rh catalyst nanoparticles. Under simulated AM 1.5G irradn., solar-to-H efficiencies of 19.3 and 18.5% are obtained in acidic and neutral electrolytes, resp. The system reaches 0.85 of the theor. limit for photoelectrochem. water splitting for the energy gap combination employed in the tandem-junction photoelectrode structure. - 9Seh, Z. W.; Kibsgaard, J.; Dickens, C. F.; Chorkendorff, I.; Nørskov, J. K.; Jaramillo, T. F. Combining Theory and Experiment in Electrocatalysis: Insights Into Materials Design. Science 2017, 355 (6321), eaad4998 DOI: 10.1126/science.aad4998
- 10Schreier, M.; Héroguel, F.; Steier, L.; Ahmad, S.; Luterbacher, J. S.; Mayer, M. T.; Luo, J.; Grätzel, M. Solar Conversion of CO2 To CO Using Earth-Abundant Electrocatalysts Prepared by Atomic Layer Modification of CuO. Nature Energy 2017, 2 (7), 17087, DOI: 10.1038/nenergy.2017.87[Crossref], [CAS], Google Scholar10https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC1cXitVeht7g%253D&md5=4efbebcfad2748d46a1361c22c435248Solar conversion of CO2 to CO using Earth-abundant electrocatalysts prepared by atomic layer modification of CuOSchreier, Marcel; Heroguel, Florent; Steier, Ludmilla; Ahmad, Shahzada; Luterbacher, Jeremy S.; Mayer, Matthew T.; Luo, Jingshan; Gratzel, MichaelNature Energy (2017), 2 (7), 17087CODEN: NEANFD; ISSN:2058-7546. (Nature Research)The solar-driven electrochem. redn. of CO2 to fuels and chems. provides a promising way for closing the anthropogenic carbon cycle. However, the lack of selective and Earth-abundant catalysts able to achieve the desired transformation reactions in an aq. matrix presents a substantial impediment as of today. Here we introduce at. layer deposition of SnO2 on CuO nanowires as a means for changing the wide product distribution of CuO-derived CO2 redn. electrocatalysts to yield predominantly CO. The activity of this catalyst towards oxygen evolution enables us to use it both as the cathode and anode for complete CO2 electrolysis. In the resulting device, the electrodes are sepd. by a bipolar membrane, allowing each half-reaction to run in its optimal electrolyte environment. Using a GaInP/GaInAs/Ge photovoltaic we achieve the solar-driven splitting of CO2 into CO and oxygen with a bifunctional, sustainable and all Earth-abundant system at an efficiency of 13.4%.
- 11Gurudayal; Beeman, J. W.; Bullock, J.; Wang, H.; Eichhorn, J.; Towle, C.; Javey, A.; Toma, F. M.; Mathews, N.; Ager, J. W., III Si Photocathode with Ag-Supported Dendritic Cu Catalyst for CO2 Reduction. Energy Environ. Sci. 2019, 12 (3), 1068– 1077, DOI: 10.1039/C8EE03547D[Crossref], [CAS], Google Scholar11https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC1MXjt1eqtbk%253D&md5=d83b4701612dc73fddb583a3e5d1da15Si photocathode with Ag-supported dendritic Cu catalyst for CO2 reductionGurudayal; Beeman, Jeffrey W.; Bullock, James; Wang, Hao; Eichhorn, Johanna; Towle, Clarissa; Javey, Ali; Toma, Francesca M.; Mathews, Nripan; Ager, Joel W.Energy & Environmental Science (2019), 12 (3), 1068-1077CODEN: EESNBY; ISSN:1754-5706. (Royal Society of Chemistry)Si photocathodes integrated with Ag-supported dendritic Cu catalysts are used to perform light-driven redn. of CO2 to C2 and C3 products in aq. soln. A back illumination geometry with an n-type Si absorber was used to permit the use of absorbing metallic catalysts. Selective carrier collection was accomplished by a p+ implantation on the illumination side and an n+ implantation followed by at. layer deposition of TiO2 on the electrolyte site. The Ag-supported dendritic Cu CO2 redn. catalyst was formed by evapn. of Ag followed by high-rate electrodeposition of Cu to form a high surface area structure. Under simulated 1 sun illumination in 0.1 M CsHCO3 satd. with CO2, the photovoltage generated by the Si (∼600 mV) enables C2 and C3 products to be produced at -0.4 vs. Texturing of both sides of the Si increases the light-limited c.d., due to reduced reflection on the illumination side, and also deceases the onset potential. Under simulated diurnal illumination conditions photocathodes maintain over 60% faradaic efficiency to hydrocarbon and oxygenate products (mainly ethylene, ethanol, propanol) for several days. After 10 days of testing, contamination from the counter electrode is obsd., which causes an increase in hydrogen prodn. This effect is mitigated by a regeneration procedure which restores the original catalyst selectivity. A tandem, self-powered CO2 redn. device was formed by coupling a Si photocathode with two series-connected semitransparent CH3NH3PbI3 perovskite solar cells, achieving an efficiency for the conversion of sunlight to hydrocarbons and oxygenates of 1.5% (3.5% for all products).
- 12Zhou, X.; Liu, R.; Sun, K.; Chen, Y.; Verlage, E.; Francis, S. A.; Lewis, N. S.; Xiang, C. Solar-Driven Reduction of 1 atm of CO2 To Formate at 10% Energy-Conversion Efficiency by Use of a TiO2-Protected III–V Tandem Photoanode in Conjunction with a Bipolar Membrane and a Pd/C Cathode. ACS Energy Lett. 2016, 1 (4), 764– 770, DOI: 10.1021/acsenergylett.6b00317[ACS Full Text], [CAS], Google Scholar12https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC28XhsV2hs77J&md5=ce1fce3fa43e3a3711ed47debe205586Solar-Driven Reduction of 1 atm of CO2 to Formate at 10% Energy-Conversion Efficiency by Use of a TiO2-Protected III-V Tandem Photoanode in Conjunction with a Bipolar Membrane and a Pd/C CathodeZhou, Xinghao; Liu, Rui; Sun, Ke; Chen, Yikai; Verlage, Erik; Francis, Sonja A.; Lewis, Nathan S.; Xiang, ChengxiangACS Energy Letters (2016), 1 (4), 764-770CODEN: AELCCP; ISSN:2380-8195. (American Chemical Society)A solar-driven CO2 redn. (CO2R) cell was constructed, consisting of a tandem GaAs/InGaP/TiO2/Ni photoanode in 1.0M KOH(aq) (pH = 13.7) to facilitate the oxygen-evolution reaction (OER), a Pd/C nanoparticle-coated Ti mesh cathode in 2.8M KHCO3(aq) (pH = 8.0) to perform the CO2R reaction, and a bipolar membrane to allow for steady-state operation of the catholyte and anolyte at different bulk pH values. At the operational c.d. of 8.5 mA cm-2, in 2.8M KHCO3(aq), the cathode exhibited <100 mV overpotential and >94% faradaic efficiency for the redn. of 1 atm of CO2(g) to formate. The anode exhibited a 320 ± 7 mV overpotential for the OER in 1.0M KOH(aq), and the bipolar membrane exhibited ∼480 mV voltage loss with minimal product crossovers and >90 and >95% selectivity for protons and hydroxide ions, resp. The bipolar membrane facilitated coupling between two electrodes and electrolytes, one for the CO2R reaction and one for the OER, that typically operate at mutually different pH values and produced a lower total cell overvoltage than known single-electrolyte CO2R systems while exhibiting ∼10% solar-to-fuels energy-conversion efficiency.
- 13Romero Cuellar, N. S.; Wiesner-Fleischer, K.; Fleischer, M.; Rucki, A.; Hinrichsen, O. Advantages of CO Over CO2 As Reactant for Electrochemical Reduction to Ethylene, Ethanol and N-Propanol on Gas Diffusion Electrodes at High Current Densities. Electrochim. Acta 2019, 307, 164– 175, DOI: 10.1016/j.electacta.2019.03.142[Crossref], [CAS], Google Scholar13https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC1MXmslOnu70%253D&md5=93b32c2a5974c6d255d14802330985f4Advantages of CO over CO2 as reactant for electrochemical reduction to ethylene, ethanol and n-propanol on gas diffusion electrodes at high current densitiesRomero Cuellar, N. S.; Wiesner-Fleischer, K.; Fleischer, M.; Rucki, A.; Hinrichsen, O.Electrochimica Acta (2019), 307 (), 164-175CODEN: ELCAAV; ISSN:0013-4686. (Elsevier Ltd.)The electrochem. conversion of CO2 to value-added chems. is a technol. gaining broader interest as society moves towards a carbon-neutral circular economy. Nonetheless, there are still several challenges to overcome before this technol. can be applied as an industrial process. In the reaction path of the electrochem. redn. of CO2 with Cu as an electrocatalyst, it is known that carbon monoxide is the key intermediate to chems. such as ethylene, ethanol, and n-propanol. However, a better understanding of the electrochem. redn. of CO is still necessary to improve selectivity and efficiency at high current densities. In this work, the electrochem. redn. of CO2 and CO towards C2 and C3 products is investigated using gas diffusion electrodes in a flow cell. Thereby the electrochem. reaction is not limited by the soly. of the feed gas in the electrolyte, and current densities of industrial relevance can be achieved. The electrodes are prepd. using com. Cu-powders consisting either of nano- or microparticles that are deposited on gas diffusion layers. Potentiostatic expts. show that with CO as the reactant, higher current densities for C2 and C3 products can be achieved at lower working electrode potentials compared to CO2 as the reactant. Galvanostatic CO electrochem. redn. at -300 mA cm-2 with Cu-nanoparticles (40-60 nm) results in a cumulative Faradaic efficiency of 89% for C2 and C3 products. This represents a two-fold increase in selectivity to ethylene and a three-fold increase towards ethanol and n-propanol compared to the selectivity obtained with CO2 as the reactant. This enhancement of selectivity for C2 and C3 products at current densities of industrial relevance with CO as reactant provides a new perspective regarding a two-step electrochem. redn. of CO2.
- 14Zhou, X.; Xiang, C. Comparative Analysis of Solar-to-Fuel Conversion Efficiency: a Direct, One-Step Electrochemical CO2 Reduction Reactor Versus a Two-Step, Cascade Electrochemical CO2 Reduction Reactor. ACS Energy Lett. 2018, 3 (8), 1892– 1897, DOI: 10.1021/acsenergylett.8b01077[ACS Full Text], [CAS], Google Scholar14https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC1cXhtlahu7rE&md5=b6d344c1806c6c31ad405ca45c6fc210Comparative Analysis of Solar-to-Fuel Conversion Efficiency: A Direct, One-Step Electrochemical CO2 Reduction Reactor versus a Two-Step, Cascade Electrochemical CO2 Reduction ReactorZhou, Xinghao; Xiang, ChengxiangACS Energy Letters (2018), 3 (8), 1892-1897CODEN: AELCCP; ISSN:2380-8195. (American Chemical Society)The solar-to-fuel conversion efficiencies of a direct, one-step reactor that electrochem. reduces CO2 to C2H6O and a two-step, cascade reactor that electrochem. reduces CO2 CO followed by a subsequent electrochem. redn. of CO to C2H6O were evaluated and compared quant. By leveraging the efficient and selective first two-electron, two proton process from CO2 to CO, the optimal solar-to-fuel conversion efficiency of the two-step reactor was higher than that of the one-step reactor at all cathodic overpotential and Faradaic efficiency combinations. The anal. shows that in some electrocatalyst performance regions with high cathodic overpotentials a relative improvement in the solar-to-fuel conversion efficiency as high as 54% can be obtained by using the two-step reactor. The alternative, two-step CO2 reactor design can provide new pathways to efficient and selective CO2 redn. to higher redn. products.
- 15Delacourt, C.; Ridgway, P. L.; Kerr, J. B.; Newman, J. Design of an Electrochemical Cell Making Syngas (CO + H2) From CO2 And H2O Reduction at Room Temperature. J. Electrochem. Soc. 2008, 155 (1), B42– B49, DOI: 10.1149/1.2801871
- 16Schulz, H. Short History and Present Trends of Fischer–Tropsch Synthesis. Appl. Catal., A 1999, 186 (1–2), 3– 12, DOI: 10.1016/S0926-860X(99)00160-X[Crossref], [CAS], Google Scholar16https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADyaK1MXmtVOkurY%253D&md5=01ee90af12a4d3a00fc0b39cbd26317bShort history and present trends of Fischer-Tropsch synthesisSchulz, H.Applied Catalysis, A: General (1999), 186 (1,2), 3-12CODEN: ACAGE4; ISSN:0926-860X. (Elsevier Science B.V.)A review, with 68 refs. Due to the large vol. of existing literature on Fischer-Tropsch (FT) synthesis, the diversity of the subject and the actually reoriented interest, it seemed indicated to write a historical sketch about the process, putting also emphasis on present trends and future options. Thus history and trends have been divided into several lines which are elaborated individually. Of course, presenting history and trends of FT synthesis on a few pages means generalizing from many individual investigations and developments and also selection of only a few citations. So I want to apologize for all the contributions to science and technol. around FT synthesis which I have missed to include into the article.
- 17Nielsen, D. U.; Hu, X.-M.; Daasbjerg, K.; Skrydstrup, T. Chemically and Electrochemically Catalysed Conversion of CO2 To CO with Follow-Up Utilization to Value-Added Chemicals. Nature Catalysis 2018, 1 (4), 244– 254, DOI: 10.1038/s41929-018-0051-3[Crossref], [CAS], Google Scholar17https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC1MXpvVyrtbc%253D&md5=beeabaa45d07b5cfb0d698e643c88949Chemically and electrochemically catalysed conversion of CO2 to CO with follow-up utilization to value-added chemicalsNielsen, Dennis U.; Hu, Xin-Ming; Daasbjerg, Kim; Skrydstrup, TroelsNature Catalysis (2018), 1 (4), 244-254CODEN: NCAACP; ISSN:2520-1158. (Nature Research)A review. Carbon dioxide is ubiquitous and a vital mol. for maintaining life on our planet. However, the ever-increasing emission of anthropogenic CO2 into our atm. has provoked dramatic climate changes. In principle, CO2 could represent an important one-carbon building block for the chem. industry, yet its high thermodn. and kinetic stability has limited its applicability to only a handful of industrial applications. On the other hand, carbon monoxide represents a more versatile reagent applied in many industrial transformations. Here we review the different methods for converting CO2 to CO with specific focus on the reverse water gas shift reaction, main element reductants, and electrochem. protocols applying homogeneous and heterogeneous catalysts. Particular emphasis is given to synthetic methods that couple the deoxygenation step with a follow-up carbonylation step for the synthesis of carbonyl-contg. mols., thus avoiding the need to handle or store this toxic but highly synthetically useful diat. gas.
- 18Lum, Y.; Ager, J. W., III. Sequential Catalysis Controls Selectivity in Electrochemical CO2 Reduction on Cu. Energy Environ. Sci. 2018, 11 (10), 2935– 2944, DOI: 10.1039/C8EE01501E[Crossref], [CAS], Google Scholar18https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC1cXht1yiu7nM&md5=d3276f3a384ae3fb6760eb13d42ba6adSequential catalysis controls selectivity in electrochemical CO2 reduction on CuLum, Yanwei; Ager, Joel W.Energy & Environmental Science (2018), 11 (10), 2935-2944CODEN: EESNBY; ISSN:1754-5706. (Royal Society of Chemistry)Electrochem. redn. of CO2 in aq. media is a strategy for sustainable prodn. of fuels and commodity chems. Cu is the only catalyst which converts CO2 to significant quantities of hydrocarbons and oxygenates. Here we demonstrate that oxygenate products can be favored over hydrocarbons by positioning a local source of CO generated by a CO producing catalyst (Au or Ag) in close proximity to a Cu catalyst. Use of a bimetallic device comprising interdigitated and independently controllable lines of Au and Cu allows the local CO concn. to be modulated. Notably, diffusional simulations show that the satn. concn. of CO can be exceeded locally. Actuating both the Au and Cu lines increases the oxygenate to ethylene ratio compared to actuating Cu only. Increasing the relative area of CO-producing Au relative to Cu also increases this ratio. These insights are translated into a second bimetallic system comprising Cu dots/lines patterned directly onto a Ag substrate, allowing for the distance between Cu and the CO generating metal to be precisely controlled. Controlling the relative areas of Ag and Cu allows for tuning of the oxygenate to ethylene ratio from 0.59 to 2.39 and an increase in oxygenate faradaic efficiency from 21.4% to 41.4%, while maintaining the selectivity to C2/C3 products in the 50-65% range. We attribute this change in selectivity to be due to an increased *CO coverage on Cu. By utilizing diffusional transport of CO to the Cu, a sequential catalysis pathway is created which allows for the control of oxygenate selectivity in aq. CO2 redn.
- 19Jouny, M.; Luc, W.; Jiao, F. High-Rate Electroreduction of Carbon Monoxide to Multi-Carbon Products. Nature Catalysis 2018, 1 (10), 748– 755, DOI: 10.1038/s41929-018-0133-2[Crossref], [CAS], Google Scholar19https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC1MXhtFGisL%252FK&md5=d0e97e6e1ae97bbde8a7c4b473693abcHigh-rate electroreduction of carbon monoxide to multi-carbon productsJouny, Matthew; Luc, Wesley; Jiao, FengNature Catalysis (2018), 1 (10), 748-755CODEN: NCAACP; ISSN:2520-1158. (Nature Research)Carbon monoxide electrolysis has previously been reported to yield enhanced multi-carbon (C2+) Faradaic efficiencies of up to ∼55%, but only at low reaction rates. This is due to the low soly. of CO in aq. electrolytes and operation in batch-type reactors. Here, we present a high-performance CO flow electrolyzer with a well controlled electrode-electrolyte interface that can reach total current densities of up to 1 A cm-2, together with improved C2+ selectivities. Computational transport modeling and isotopic C18O redn. expts. suggest that the enhanced activity is due to a higher surface pH under CO redn. conditions, which facilitates the prodn. of acetate. At optimal operating conditions, we achieve a C2+ Faradaic efficiency of ∼91% with a C2+ partial c.d. over 630 mA cm-2. Further investigations show that maintaining an efficient triple-phase boundary at the electrode-electrolyte interface is the most crit. challenge in achieving a stable CO/CO2 electrolysis process at high rates.
- 20Welch, A. J.; DuChene, J. S.; Tagliabue, G.; Davoyan, A.; Cheng, W.-H.; Atwater, H. A. Nanoporous Gold as a Highly Selective and Active Carbon Dioxide Reduction Catalyst. ACS Appl. Energy Mater. 2019, 2 (1), 164– 170, DOI: 10.1021/acsaem.8b01570[ACS Full Text], [CAS], Google Scholar20https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC1cXisF2lurzM&md5=1c0bba179a63dc7ac0d3af6d2bd1eb5fNanoporous Gold as a Highly Selective and Active Carbon Dioxide Reduction CatalystWelch, Alex J.; DuChene, Joseph S.; Tagliabue, Giulia; Davoyan, Artur; Cheng, Wen-Hui; Atwater, Harry A.ACS Applied Energy Materials (2019), 2 (1), 164-170CODEN: AAEMCQ; ISSN:2574-0962. (American Chemical Society)Electrochem. conversion of CO2 into useful chems. is a promising approach for transforming CO2 into sustainably produced fuels and/or chem. feedstocks for industrial synthesis. The authors report that nanoporous Au (np-Au) films, with pore sizes ranging from 10 to 30 nm, represent promising electrocatalytic architectures for the CO2 redn. reaction (CO2RR) due to their large electrochem. active surface area, relative abundance of grain boundaries, and ability to support pH gradients inside the nanoporous network. Electrochem. studies show that np-Au films support partial current densities for the conversion of CO2 to CO >6 mA cm-2 at a faradaic efficiency of ∼99% in aq. electrolytes (50 mM K2CO3 satd. with CO2). Also, np-Au films are able to maintain faradaic efficiency >80% for CO prodn. over prolonged periods of continuous operation (110 h). Electrocatalytic expts. at different electrolyte concns. demonstrate that the pore diam. of nanoporous cathodes represents a crit. parameter for creating and controlling local pH gradients inside the porous network of metal ligaments. These results demonstrate the merits of nanoporous metal films for the CO2RR and offer an interesting architecture for highly selective electrocatalysis capable of sustaining high catalytic currents over prolonged periods.
- 21Chen, Y.; Li, C. W.; Kanan, M. W. Aqueous CO2 Reduction at Very Low Overpotential on Oxide-Derived Au Nanoparticles. J. Am. Chem. Soc. 2012, 134 (49), 19969– 19972, DOI: 10.1021/ja309317u[ACS Full Text], [CAS], Google Scholar21https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC38Xhslalu7rN&md5=bc3f0624d8d46f9e16aca5d1a0f66420Aqueous CO2 Reduction at Very Low Overpotential on Oxide-Derived Au NanoparticlesChen, Yihong; Li, Christina W.; Kanan, Matthew W.Journal of the American Chemical Society (2012), 134 (49), 19969-19972CODEN: JACSAT; ISSN:0002-7863. (American Chemical Society)Carbon dioxide redn. is an essential component of many prospective technologies for the renewable synthesis of carbon-contg. fuels. Known catalysts for this reaction generally suffer from low energetic efficiency, poor product selectivity, and rapid deactivation. It is shown that the redn. of thick Au oxide films results in the formation of Au nanoparticles (oxide-derived Au) that exhibit highly selective CO2 redn. to CO in water at overpotentials as low as 140 mV and retain their activity for at least 8 h. Under identical conditions, polycryst. Au electrodes and several other nanostructured Au electrodes prepd. via alternative methods require at least 200 mV of addnl. overpotential to attain comparable CO2 redn. activity and rapidly lose their activity. Electrokinetic studies indicate that the improved catalysis is linked to dramatically increased stabilization of the CO2•- intermediate on the surfaces of the oxide-derived Au electrodes.
- 22Hatsukade, T.; Kuhl, K. P.; Cave, E. R.; Abram, D. N.; Jaramillo, T. F. Insights Into the Electrocatalytic Reduction of CO2 On Metallic Silver Surfaces. Phys. Chem. Chem. Phys. 2014, 16 (27), 13814– 13819, DOI: 10.1039/C4CP00692E[Crossref], [PubMed], [CAS], Google Scholar22https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC2cXhtVantrjM&md5=639d007871b7b111fbc30803d23add94Insights into the electrocatalytic reduction of CO2 on metallic silver surfacesHatsukade, Toru; Kuhl, Kendra P.; Cave, Etosha R.; Abram, David N.; Jaramillo, Thomas F.Physical Chemistry Chemical Physics (2014), 16 (27), 13814-13819CODEN: PPCPFQ; ISSN:1463-9076. (Royal Society of Chemistry)The electrochem. redn. of CO2 could allow for a sustainable process by which renewable energy from wind and solar are used directly in the prodn. of fuels and chems. In this work we investigated the potential dependent activity and selectivity of the electrochem. redn. of CO2 on metallic silver surfaces under ambient conditions. Our results deepen our understanding of the surface chem. and provide insight into the factors important to designing better catalysts for the reaction. The high sensitivity of our exptl. methods for identifying and quantifying products of reaction allowed for the observation of six redn. products including CO and hydrogen as major products and formate, methane, methanol, and ethanol as minor products. By quantifying the potential-dependent behavior of all products, we provide insights into kinetics and mechanisms at play, in particular involving the prodn. of hydrocarbons and alcs. on catalysts with weak CO binding energy as well as the formation of a C-C bond required to produce ethanol.
- 23Asadi, M.; Kim, K.; Liu, C.; Addepalli, A. V.; Abbasi, P.; Yasaei, P.; Phillips, P.; Behranginia, A.; Cerrato, J. M.; Haasch, R.; Zapol, P.; Kumar, B.; Klie, R. F.; Abiade, J.; Curtiss, L. A.; Salehi-Khojin, A. Nanostructured Transition Metal Dichalcogenide Electrocatalysts for CO2 Reduction in Ionic Liquid. Science 2016, 353 (6298), 467– 470, DOI: 10.1126/science.aaf4767[Crossref], [PubMed], [CAS], Google Scholar23https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC28Xht1ehtL7M&md5=b2a2c08639c77e7ca8afd6c4c1a08bd4Nanostructured transition metal dichalcogenide electrocatalysts for CO2 reduction in ionic liquidAsadi, Mohammad; Kim, Kibum; Liu, Cong; Addepalli, Aditya Venkata; Abbasi, Pedram; Yasaei, Poya; Phillips, Patrick; Behranginia, Amirhossein; Cerrato, Jose M.; Haasch, Richard; Zapol, Peter; Kumar, Bijandra; Klie, Robert F.; Abiade, Jeremiah; Curtiss, Larry A.; Salehi-Khojin, AminScience (Washington, DC, United States) (2016), 353 (6298), 467-470CODEN: SCIEAS; ISSN:0036-8075. (American Association for the Advancement of Science)Conversion of carbon dioxide (CO2) into fuels is an attractive soln. to many energy and environmental challenges. However, the chem. inertness of CO2 renders many electrochem. and photochem. conversion processes inefficient. A transition metal dichalcogenide nanoarchitecture is reported for catalytic electrochem. CO2 conversion to carbon monoxide (CO) in an ionic liq. It is found that tungsten diselenide nanoflakes show a c.d. of 18.95 mA per square centimeter, CO faradaic efficiency of 24%, and CO formation turnover frequency of 0.28 per s at a low overpotential of 54 mV. The catalyst is also applied in a light-harvesting artificial leaf platform that concurrently oxidized water in the absence of any external potential.
- 24Asadi, M.; Kumar, B.; Behranginia, A.; Rosen, B. A.; Baskin, A.; Repnin, N.; Pisasale, D.; Phillips, P.; Zhu, W.; Haasch, R.; Klie, R. F.; Král, P.; Abiade, J.; Salehi-Khojin, A. Robust Carbon Dioxide Reduction on Molybdenum Disulphide Edges.. Nat. Commun. 2014, 5, 4470, DOI: 10.1038/ncomms5470[Crossref], [PubMed], [CAS], Google Scholar24https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC2cXhvF2lsr%252FF&md5=81f74b1c1a0ee5987742eb6cbfb03c9eRobust carbon dioxide reduction on molybdenum disulphide edgesAsadi, Mohammad; Kumar, Bijandra; Behranginia, Amirhossein; Rosen, Brian A.; Baskin, Artem; Repnin, Nikita; Pisasale, Davide; Phillips, Patrick; Zhu, Wei; Haasch, Richard; Klie, Robert F.; Kral, Petr; Abiade, Jeremiah; Salehi-Khojin, AminNature Communications (2014), 5 (), 4470CODEN: NCAOBW; ISSN:2041-1723. (Nature Publishing Group)Electrochem. redn. of carbon dioxide has been recognized as an efficient way to convert carbon dioxide to energy-rich products. Noble metals (for example, gold and silver) have been demonstrated to reduce carbon dioxide at moderate rates and low overpotentials. Nevertheless, the development of inexpensive systems with an efficient carbon dioxide redn. capability remains a challenge. Here we identify molybdenum disulfide as a promising cost-effective substitute for noble metal catalysts. We uncover that molybdenum disulfide shows superior carbon dioxide redn. performance compared with the noble metals with a high c.d. and low overpotential (54 mV) in an ionic liq. Scanning transmission electron microscopy anal. and first principle modeling reveal that the molybdenum-terminated edges of molybdenum disulfide are mainly responsible for its catalytic performance due to their metallic character and a high d-electron d. This is further exptl. supported by the carbon dioxide redn. performance of vertically aligned molybdenum disulfide.
- 25Cheng, T.; Huang, Y.; Xiao, H.; Goddard, W. A. Predicted Structures of the Active Sites Responsible for the Improved Reduction of Carbon Dioxide by Gold Nanoparticles. J. Phys. Chem. Lett. 2017, 8 (14), 3317– 3320, DOI: 10.1021/acs.jpclett.7b01335[ACS Full Text], [CAS], Google Scholar25https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC2sXhtFSnu7vN&md5=3108377ea91d863955ec4e554eb49807Predicted Structures of the Active Sites Responsible for the Improved Reduction of Carbon Dioxide by Gold NanoparticlesCheng, Tao; Huang, Yufeng; Xiao, Hai; Goddard, William A.Journal of Physical Chemistry Letters (2017), 8 (14), 3317-3320CODEN: JPCLCD; ISSN:1948-7185. (American Chemical Society)Gold (Au) nanoparticles (NPs) are known exptl. to reduce carbon dioxide (CO2) to carbon monoxide (CO), with far superior performance to Au foils. To obtain guidance in designing improved CO2 catalysts, we want to understand the nature of the active sites on Au NPs. Here, we employed multiscale atomistic simulations to computationally synthesize and characterize a 10 nm thick Au NP on a carbon nanotube (CNT) support, and then we located active sites from quantum mechanics (QM) calcns. on 269 randomly selected sites. The std. scaling relation is that the formation energy of *COOH (ΔE*COOH) is proportional to the binding energy of *CO (Ebinding*CO); therefore, decreasing ΔE*COOH to boost the CO2 redn. reaction (CO2RR) causes an increase of Ebinding*CO that retards CO2RR. We show that the NPs have superior CO2RR because there are many sites at the twin boundaries that significantly break this scaling relation.
- 26Singh, M. R.; Kwon, Y.; Lum, Y.; Ager, J. W., III; Bell, A. T. Hydrolysis of Electrolyte Cations Enhances the Electrochemical Reduction of CO2 Over Ag and Cu. J. Am. Chem. Soc. 2016, 138 (39), 13006– 13012, DOI: 10.1021/jacs.6b07612[ACS Full Text], [CAS], Google Scholar26https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC28XhsFWjur3P&md5=e1a7c5a8f8ee0451fe834febe8b1d123Hydrolysis of Electrolyte Cations Enhances the Electrochemical Reduction of CO2 over Ag and CuSingh, Meenesh R.; Kwon, Youngkook; Lum, Yanwei; Ager, Joel W.; Bell, Alexis T.Journal of the American Chemical Society (2016), 138 (39), 13006-13012CODEN: JACSAT; ISSN:0002-7863. (American Chemical Society)Electrolyte cation size is known to influence the electrochem. redn. of CO2 over metals; however, a satisfactory explanation for this phenomenon was not developed. The authors report here that these effects can be attributed to a previously unrecognized consequence of cation hydrolysis occurring in the vicinity of the cathode. With increasing cation size, the pKa for cation hydrolysis decreases and is sufficiently low for hydrated K+, Rb+, and Cs+ to serve as buffering agents. Buffering lowers the pH near the cathode, increasing the local concn. of dissolved CO2. The consequences of these changes are an increase in cathode activity, a decrease in faradaic efficiencies for H2 and CH4, and an increase in faradaic efficiencies for CO, C2H4, and EtOH, in full agreement with exptl. observations for CO2 redn. over Ag and Cu.
- 27Liu, M.; Pang, Y.; Zhang, B.; De Luna, P.; Voznyy, O.; Xu, J.; Zheng, X.; Dinh, C. T.; Fan, F.; Cao, C.; de Arquer, F. P. G.; Safaei, T. S.; Mepham, A.; Klinkova, A.; Kumacheva, E.; Filleter, T.; Sinton, D.; Kelley, S. O.; Sargent, E. H. Enhanced Electrocatalytic CO2 Reduction via Field-Induced Reagent Concentration. Nature 2016, 537 (7620), 382– 386, DOI: 10.1038/nature19060[Crossref], [PubMed], [CAS], Google Scholar27https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC28Xht1yqsbzO&md5=11d8dcf8010b105587ba93d7035bc657Enhanced electrocatalytic CO2 reduction via field-induced reagent concentrationLiu, Min; Pang, Yuanjie; Zhang, Bo; De Luna, Phil; Voznyy, Oleksandr; Xu, Jixian; Zheng, Xueli; Dinh, Cao Thang; Fan, Fengjia; Cao, Changhong; Garcia de Arquer, F. Pelayo; Safaei, Tina Saberi; Mepham, Adam; Klinkova, Anna; Kumacheva, Eugenia; Filleter, Tobin; Sinton, David; Kelley, Shana O.; Sargent, Edward H.Nature (London, United Kingdom) (2016), 537 (7620), 382-386CODEN: NATUAS; ISSN:0028-0836. (Nature Publishing Group)Electrochem. redn. of CO2 (CO2) to CO (CO) is the 1st step in the synthesis of more complex C-based fuels and feedstocks using renewable electricity. Unfortunately, the reaction suffers from slow kinetics owing to the low local concn. of CO2 surrounding typical CO2 redn. reaction catalysts. Alkali metal cations are known to overcome this limitation through noncovalent interactions with adsorbed reagent species, but the effect is restricted by the soly. of relevant salts. Large applied electrode potentials can also enhance CO2 adsorption, but this comes at the cost of increased H (H2) evolution. Here we report that nanostructured electrodes produce, at low applied overpotentials, local high elec. fields that conc. electrolyte cations, which in turn leads to a high local concn. of CO2 close to the active CO2 redn. reaction surface. Simulations reveal 10-fold higher elec. fields assocd. with metallic nm-sized tips compared to quasi-planar electrode regions, and measurements using Au nanoneedles confirm a field-induced reagent concn. that enables the CO2 redn. reaction to proceed with a geometric c.d. for CO of 22 mA per square centimeter at -0.35 V (overpotential of 0.24 V). This performance surpasses by an order of magnitude the performance of the best Au nanorods, nanoparticles and oxide-derived noble metal catalysts. Similarly designed Pd nanoneedle electrocatalysts produce formate with a faradaic efficiency of >90 per cent and an unprecedented geometric c.d. for formate of 10 mA per square centimeter at -0.2 V, demonstrating the wider applicability of the field-induced reagent concn. concept.
- 28Varela, A. S.; Kroschel, M.; Reier, T.; Strasser, P. Controlling the Selectivity of CO2 Electroreduction on Copper: the Effect of the Electrolyte Concentration and the Importance of the Local pH. Catal. Today 2016, 260, 8– 13, DOI: 10.1016/j.cattod.2015.06.009[Crossref], [CAS], Google Scholar28https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC2MXhtFeitrrE&md5=3af2d3ae954b9ce2d779503eb6bca663Controlling the selectivity of CO2 electroreduction on copper: The effect of the electrolyte concentration and the importance of the local pHVarela, Ana Sofia; Kroschel, Matthias; Reier, Tobias; Strasser, PeterCatalysis Today (2016), 260 (), 8-13CODEN: CATTEA; ISSN:0920-5861. (Elsevier B.V.)The activity and selectivity of Cu during the CO2 electrochem. redn. can be tuned by changing the concn. of the bicarbonate electrolyte. Comparing the abs. formation rate and faradaic selectivity of H2, CH4, CO, and C2H4 as a function of the applied electrode potential, variations in the bulk buffer capacities of the electrolyte have substantial impact on abs. product formation rates and relative faradaic selectivity. High concns. of bicarbonate improve the overall faradaic CO2 electroredn. activity, largely due to higher abs. formation rates of H2 and CH4. In lower-concd. bicarbonate electrolytes with their lower overall activity, the selectivity toward ethylene was drastically enhanced. Following earlier theor. work, the authors hypothesize the pH near the Cu electrode interface to largely account for the obsd. effects: dild. KHCO3 solns. allow for more alk. local pH values during CO2 electroredn. The authors' study highlights the controlling role of the interfacial pH on the product distribution during CO2 redn. over a wide electrode potential range.
- 29Verma, S.; Lu, X.; Ma, S.; Masel, R. I.; Kenis, P. J. A. The Effect of Electrolyte Composition on the Electroreduction of CO2 To CO on Ag Based Gas Diffusion Electrodes. Phys. Chem. Chem. Phys. 2016, 18 (10), 7075– 7084, DOI: 10.1039/C5CP05665A[Crossref], [PubMed], [CAS], Google Scholar29https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC2MXhvVygs7rF&md5=d489d730587522ee37bf3c28c511c77aThe effect of electrolyte composition on the electroreduction of CO2 to CO on Ag based gas diffusion electrodesVerma, Sumit; Lu, Xun; Ma, Sichao; Masel, Richard I.; Kenis, Paul J. A.Physical Chemistry Chemical Physics (2016), 18 (10), 7075-7084CODEN: PPCPFQ; ISSN:1463-9076. (Royal Society of Chemistry)The electroredn. of CO2 to C1-C2 chems. can be a potential strategy for using CO2 as a C feedstock. The authors study the effect of electrolytes on the electroredn. of CO2 to CO on Ag based gas diffusion electrodes. Electrolyte concn. was found to play a major role in the process for the electrolytes (KOH, KCl, and KHCO3) studied here. Several fold improvements in partial current densities of CO (jCO) were obsd. on moving from 0.5 M to 3.0 M electrolyte soln. independent of the nature of the anion. jCO values ≤440 mA cm-2 with an energy efficiency (EE) of ≈ 42% and 230 mA cm-2 with EE ≈ 54% were obsd. when using 3.0 M KOH. Electrochem. impedance spectroscopy showed that both the charge transfer resistance (Rct) and the cell resistance (Rcell) decreased on moving from a 0.5 M to a 3.0 M KOH electrolyte. Anions play an important role with respect to reducing the onset potential of CO in the order OH- (-0.13 V vs. RHE) < HCO3- (-0.46 V vs. RHE) < Cl- (-0.60 V vs. RHE). A decrease in Rct upon increasing electrolyte concn. and the effect of anions on the cathode can be explained by an interplay of different interactions in the elec. double layer that can either stabilize or destabilize the rate limiting CO2̇ - radical. EMIM based ionic liqs. and 1 : 2 choline Cl urea based deep eutectic solvents (DESs) were used for CO2 capture but exhibit low cond. Here, the authors study if the addn. of KCl to such solns. can improve cond. and hence jCO. Electrolytes contg. KCl in combination with EMIM Cl, choline Cl, or DESs showed a two to three fold improvement in jCO in comparison to those without KCl. Using such mixts. can be a strategy for integrating the process of CO2 capture with CO2 conversion.
- 30Higgins, D.; Hahn, C.; Xiang, C.; Jaramillo, T. F.; Weber, A. Z. Gas-Diffusion Electrodes for Carbon Dioxide Reduction: a New Paradigm. ACS Energy Lett. 2019, 4 (1), 317– 324, DOI: 10.1021/acsenergylett.8b02035[ACS Full Text], [CAS], Google Scholar30https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC1cXisFehur%252FK&md5=4eb4228156f69de1cf3f61a7011b36a9Gas-Diffusion Electrodes for Carbon Dioxide Reduction: A New ParadigmHiggins, Drew; Hahn, Christopher; Xiang, Chengxiang; Jaramillo, Thomas F.; Weber, Adam Z.ACS Energy Letters (2019), 4 (1), 317-324CODEN: AELCCP; ISSN:2380-8195. (American Chemical Society)A review. Significant advances have been made in recent years discovering new electrocatalysts and developing a fundamental understanding of electrochem. CO2 redn. processes. This field has progressed to the point that efforts can now focus on translating this knowledge toward the development of practical CO2 electrolyzers, which have the potential to replace conventional petrochem. processes as a sustainable route to produce fuels and chems. In this Perspective, we take a crit. look at the progress in incorporating electrochem. CO2 redn. catalysts into practical device architectures that operate using vapor-phase CO2 reactants, thereby overcoming intrinsic limitations of aq.-based systems. Performance comparison is made between state-of-the-art CO2 electrolyzers and com. H2O electrolyzers-a well-established technol. that provides realistic performance targets. Beyond just higher rates, vapor-fed reactors represent new paradigms for unprecedented control of local reaction conditions, and we provide a perspective on the challenges and opportunities for generating fundamental knowledge and achieving technol. progress toward the development of practical CO2 electrolyzers.
- 31Singh, M. R.; Papadantonakis, K.; Xiang, C.; Lewis, N. S. An Electrochemical Engineering Assessment of the Operational Conditions and Constraints for Solar-Driven Water-Splitting Systems at Near-Neutral pH. Energy Environ. Sci. 2015, 8 (9), 2760– 2767, DOI: 10.1039/C5EE01721A[Crossref], [CAS], Google Scholar31https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC2MXhtFSksLzN&md5=b5918f12163160e02f793d28857d186bAn electrochemical engineering assessment of the operational conditions and constraints for solar-driven water-splitting systems at near-neutral pHSingh, Meenesh R.; Papadantonakis, Kimberly; Xiang, Chengxiang; Lewis, Nathan S.Energy & Environmental Science (2015), 8 (9), 2760-2767CODEN: EESNBY; ISSN:1754-5706. (Royal Society of Chemistry)The soln. transport losses in a one-dimensional solar-driven water-splitting cell that operates in either concd. acid, dil. acid, or buffered near-neutral pH electrolytes have been evaluated using a math. model that accounts for diffusion, migration and convective transport, as well as for bulk electrochem. reactions in the electrolyte. The Ohmic resistance loss, the Nernstian potential loss assocd. with pH gradients at the surface of the electrode, and electrodialysis in different electrolytes were assessed quant. in a stagnant cell as well as in a bubble-convected cell, in which convective mixing occurred due to product-gas evolution. In a stagnant cell that did not have convective mixing, small limiting current densities (<3 mA cm-2) and significant polarization losses derived from pH gradients were present in dil. acid as well as in near-neutral pH buffered electrolytes. In contrast, bubble-convected cells exhibited a significant increase in the limiting c.d., and a significant redn. of the concn. overpotentials. In a bubble-convected cell, minimal soln. transport losses were present in membrane-free cells, in either buffered electrolytes or in unbuffered solns. with pH ≤ 1. However, membrane-free cells lack a mechanism for product-gas sepn., presenting significant practical and engineering impediments to the deployment of such systems. To produce an intrinsically safe cell, an ion-exchange membrane was incorporated into the cell. The accompanying soln. losses, esp. the pH gradients at the electrode surfaces, were modeled and simulated for such a system. Hence this work describes the general conditions under which intrinsically safe, efficient solar-driven water-splitting cells can be operated.
- 32Lobaccaro, P.; Singh, M. R.; Clark, E. L.; Kwon, Y.; Bell, A. T.; Ager, J. W., III. Effects of Temperature and Gas–Liquid Mass Transfer on the Operation of Small Electrochemical Cells for the Quantitative Evaluation of CO2 Reduction Electrocatalysts. Phys. Chem. Chem. Phys. 2016, 18 (38), 26777– 26785, DOI: 10.1039/C6CP05287H[Crossref], [PubMed], [CAS], Google Scholar32https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC28XhsVKitr3F&md5=b9b3f9bf7bbf205dddb69272df3aa009Effects of temperature and gas-liquid mass transfer on the operation of small electrochemical cells for the quantitative evaluation of CO2 reduction electrocatalystsLobaccaro, Peter; Singh, Meenesh R.; Clark, Ezra Lee; Kwon, Youngkook; Bell, Alexis T.; Ager, Joel W.Physical Chemistry Chemical Physics (2016), 18 (38), 26777-26785CODEN: PPCPFQ; ISSN:1463-9076. (Royal Society of Chemistry)In the last few years, there has been increased interest in electrochem. CO2 redn. (CO2R). Many exptl. studies employ a membrane sepd., electrochem. cell with a mini H-cell geometry to characterize CO2R catalysts in aq. soln. This type of electrochem. cell is a mini-chem. reactor and it is important to monitor the reaction conditions within the reactor to ensure that they are const. throughout the study. We show that operating cells with high catalyst surface area to electrolyte vol. ratios (S/V) at high current densities can have subtle consequences due to the complexity of the phys. phenomena taking place on electrode surfaces during CO2R, particularly as they relate to the cell temp. and bulk electrolyte CO2 concn. Both effects were evaluated quant. in high S/V cells using Cu electrodes and a bicarbonate buffer electrolyte. Electrolyte temp. is a function of the current/total voltage passed through the cell and the cell geometry. Even at a very high c.d., 20 mA cm-2, the temp. increase was less than 4 °C and a decrease of <10% in the dissolved CO2 concn. is predicted. In contrast, limits on the CO2 gas-liq. mass transfer into the cells produce much larger effects. By using the pH in the cell to measure the CO2 concn., significant undersatn. of CO2 is obsd. in the bulk electrolyte, even at more modest current densities of 10 mA cm-2. Undersatn. of CO2 produces large changes in the faradaic efficiency obsd. on Cu electrodes, with H2 prodn. becoming increasingly favored. We show that the size of the CO2 bubbles being introduced into the cell is crit. for maintaining the equil. CO2 concn. in the electrolyte, and we have designed a high S/V cell that is able to maintain the near-equil. CO2 concn. at current densities up to 15 mA cm-2.
- 33Weng, L.-C.; Bell, A. T.; Weber, A. Z. Towards Membrane-Electrode Assembly Systems for CO2 Reduction: a Modeling Study. Energy Environ. Sci. 2019, 12 (6), 1950– 1968, DOI: 10.1039/C9EE00909D[Crossref], [CAS], Google Scholar33https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC1MXptFKhu70%253D&md5=423c940774b5d165b0c50a0a0dfb6256Towards membrane-electrode assembly systems for CO2 reduction: a modeling studyWeng, Lien-Chun; Bell, Alexis T.; Weber, Adam Z.Energy & Environmental Science (2019), 12 (6), 1950-1968CODEN: EESNBY; ISSN:1754-5706. (Royal Society of Chemistry)Membrane-electrode assemblies (MEAs) are an attractive cell design for the electrochem. redn. of CO2 because they exhibit low ohmic loss and high energy efficiency. We describe here the development and application of a multiphysics model to investigate the fundamental limitations of two MEA designs: one with gaseous feeds at both the anode and cathode (full-MEA), and the other with an aq. anode feed (KHCO3 or KOH exchange soln.) and a gaseous cathode feed (exchange-MEA). The total c.d. for the three cases follows the order: KOH-MEA > KHCO3-MEA > full-MEA. This trend is established by examg. the distribution of the applied voltage. We show that the main charge-carrying species are carbonate anions for an MEA that uses an anion-exchange membrane (AEM). The amt. of CO2 consumed but not converted to CO decreases with increasing current densities above 100 mA cm-2 for a full-MEA, but converges to 50% for exchange-MEAs. The full-MEA becomes limited by ohmic resistance as the membrane dehydrates with increasing cell temp., and eventually becomes limited due to water mass transport. The exchange-MEAs can maintain membrane hydration and the local ion concn. at the anode, but are limited by salt pptn. at the cathode, as well as a higher tendency to flood. Finally, we explore the effects of temp. and discuss the possibility of increasing water supply to the full-MEA to improve its performance at elevated temps. The MEA model and the understanding of MEA performance for the electrochem. redn. of CO2 presented in this study should help guide the design of next-generation CO2 redn. cells.
- 34Song, J. T.; Song, H.; Kim, B.; Oh, J. Towards Higher Rate Electrochemical CO2 Conversion: From Liquid-Phase to Gas-Phase Systems. Catalysts 2019, 9 (3), 224, DOI: 10.3390/catal9030224[Crossref], [CAS], Google Scholar34https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC1MXotlGjtrw%253D&md5=ec441ee8bd7ad07f1d52c09e4259e31eTowards higher rate electrochemical CO2 conversion: from liquid-phase to gas-phase systemsSong, Jun Tae; Song, Hakhyeon; Kim, Beomil; Oh, JihunCatalysts (2019), 9 (3), 224/1-224/25CODEN: CATACJ; ISSN:2073-4344. (MDPI AG)Electrochem. CO2 conversion offers a promising route for value-added products such as formate, carbon monoxide, and hydrocarbons. As a result of the highly required overpotential for CO2 redn., researchers have extensively studied the development of catalyst materials in a typical H-type cell, utilizing a dissolved CO2 reactant in the liq. phase. However, the low CO2 soly. in an aq. soln. has critically limited productivity, thereby hindering its practical application. In efforts to realize com. available CO2 conversion, gas-phase reactor systems have recently attracted considerable attention. Although the achieved performance to date reflects a high feasibility, further development is still required in order for a well-established technol. Accordingly, this review aims to promote the further study of gas-phase systems for CO2 redn., by generally examg. some previous approaches from liq.-phase to gas-phase systems. Finally, we outline major challenges, with significant lessons for practical CO2 conversion systems.
- 35Li, J.; Chen, G.; Zhu, Y.; Liang, Z.; Pei, A.; Wu, C.-L.; Wang, H.; Lee, H. R.; Liu, K.; Chu, S.; Cui, Y. Efficient Electrocatalytic CO2 Reduction on a Three-Phase Interface. Nature Catalysis 2018, 1 (8), 592– 600, DOI: 10.1038/s41929-018-0108-3[Crossref], [CAS], Google Scholar35https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC1MXhtFGisb7I&md5=d2fb2bf7cb72e2e88d22a4fdbd8d2700Efficient electrocatalytic CO2 reduction on a three-phase interfaceLi, Jun; Chen, Guangxu; Zhu, Yangying; Liang, Zheng; Pei, Allen; Wu, Chun-Lan; Wang, Hongxia; Lee, Hye Ryoung; Liu, Kai; Chu, Steven; Cui, YiNature Catalysis (2018), 1 (8), 592-600CODEN: NCAACP; ISSN:2520-1158. (Nature Research)Electrochem. CO2 redn. is a crit. approach to reducing the globally accelerating CO2 emission and generating value-added products. Despite great efforts to optimize catalyst activity and selectivity, facilitating the catalyst accessibility to high CO2 concns. while maintaining electrode durability remains a significant challenge. Here, we designed a catalytic system that mimics the alveolus structure in mammalian lungs with high gas permeability but very low water diffusibility, enabling an array of three-phase catalytic interfaces. Flexible, hydrophobic, nanoporous polyethylene membranes with high gas permeability were used to enable efficient CO2 access and a high local alky. on the catalyst surface at different CO2 flow rates. Such an alveolus-mimicking structure generates a high CO prodn. Faradaic efficiency of 92% and excellent geometric current densities of CO prodn. (25.5 mA cm-2) at -0.6 V vs. the reversible hydrogen electrode, with a very thin catalyst thickness of 20-80 nm.
- 36Weng, L.-C.; Bell, A. T.; Weber, A. Z. Modeling Gas-Diffusion Electrodes for CO2 Reduction. Phys. Chem. Chem. Phys. 2018, 20, 16973– 16984, DOI: 10.1039/C8CP01319E[Crossref], [PubMed], [CAS], Google Scholar36https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC1cXhtVKntb%252FI&md5=d5beb1df920eac710ede8615667d7e11Modeling gas-diffusion electrodes for CO2 reductionWeng, Lien-Chun; Bell, Alexis T.; Weber, Adam Z.Physical Chemistry Chemical Physics (2018), 20 (25), 16973-16984CODEN: PPCPFQ; ISSN:1463-9076. (Royal Society of Chemistry)CO2 redn. conducted in electrochem. cells with planar electrodes immersed in an aq. electrolyte is severely limited by mass transport across the hydrodynamic boundary layer. This limitation can be minimized by use of vapor-fed, gas-diffusion electrodes (GDEs), enabling current densities that are almost two orders of magnitude greater at the same applied cathode overpotential than what is achievable with planar electrodes in an aq. electrolyte. The addn. of porous cathode layers, however, introduces a no. of parameters that need to be tuned in order to optimize the performance of the GDE cell. In this work, we develop a multiphysics model for gas diffusion electrodes for CO2 redn. and used it to investigate the interplay between species transport and electrochem. reaction kinetics. The model demonstrates how the local environment near the catalyst layer, which is a function of the operating conditions, affects cell performance. We also examine the effects of catalyst layer hydrophobicity, loading, porosity, and electrolyte flowrate to help guide exptl. design of vapor-fed CO2 redn. cells.
- 37Cook, R. L.; MacDuff, R. C.; Sammells, A. F. High Rate Gas Phase CO2 Reduction to Ethylene and Methane Using Gas Diffusion Electrodes. J. Electrochem. Soc. 1990, 137 (2), 607– 608, DOI: 10.1149/1.2086515[Crossref], [CAS], Google Scholar37https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADyaK3cXhsVOkurw%253D&md5=0fbbb2a641aaed5ebaf1cd904242cf33High rate gas phase carbon dioxide reduction to ethylene and methane using gas diffusion electrodesCook, Ronald L.; MacDuff, Robert C.; Sammells, Anthony F.Journal of the Electrochemical Society (1990), 137 (2), 607-8CODEN: JESOAN; ISSN:0013-4651.Gas phase redn. of CO2 was studied at gas diffusion electrodes made from Cu gauge with C black and Teflon with and without supported Cu. Cu is the active site for CO2 redn. Ethylene was the dominant redn. product. Faradaic efficiencies of 71.3% were obtained for gas phase redn. of CO2 to hydrocarbon at c.d. >0.5 A/cm2 using the gas diffusion electrode.
- 38Hori, Y.; Ito, H.; Okano, K.; Nagasu, K.; Sato, S. Silver-Coated Ion Exchange Membrane Electrode Applied to Electrochemical Reduction of Carbon Dioxide. Electrochim. Acta 2003, 48 (18), 2651– 2657, DOI: 10.1016/S0013-4686(03)00311-6[Crossref], [CAS], Google Scholar38https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BD3sXltFCmsb4%253D&md5=65df35bb823ad0fbf9820b1cbd560e68Silver-coated ion exchange membrane electrode applied to electrochemical reduction of carbon dioxideHori, Y.; Ito, H.; Okano, K.; Nagasu, K.; Sato, S.Electrochimica Acta (2003), 48 (18), 2651-2657CODEN: ELCAAV; ISSN:0013-4686. (Elsevier Science B.V.)Silver-coated ion exchange membrane electrodes (solid polymer electrolyte, SPE) were prepd. by electroless deposition of silver onto ion exchange membranes. The SPE electrodes were used for carbon dioxide (CO2) redn. with 0.2 M K2SO4 as the electrolyte with a platinum plate (Pt) for the counterelectrode. In an SPE electrode system prepd. from a cation exchange membrane (CEM), the surface of the SPE was partly ruptured during CO2 redn., and the reaction was rapidly suppressed. SPE electrodes made of an anion exchange membrane (SPE/AEM) sustained redn. of CO2 to CO for more than 2 h, whereas, the electrode potential shifted neg. during the electrolysis. The reaction is controlled by the diffusion of CO2 through the metal layer of the SPE electrode at high c.d. Ultrasonic radiation, applied to the prepn. of SPE/AEM, was effective to improve the electrode properties, enhancing the electrolysis current of CO2 redn. Observation by a scanning electron microscope (SEM) showed that the electrode metal layer became more porous by the ultrasonic radiation treatment. The partial c.d. of CO2 redn. by SPE/AEM amounted to 60 mA cm-2, i.e. three times the upper limit of the conventional electrolysis by a plate electrode. Application of SPE device may contribute to an advancement of CO2 fixation at ambient temp. and pressure.
- 39CRC Handbook of Chemistry and Physics; Lide, D. R., Ed.; New York, 2003.
- 40Ripatti, D. S.; Veltman, T. R.; Kanan, M. W. Carbon Monoxide Gas Diffusion Electrolysis That Produces Concentrated C2 Products with High Single-Pass Conversion. Joule 2019, 3 (1), 240– 256, DOI: 10.1016/j.joule.2018.10.007[Crossref], [CAS], Google Scholar40https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC1MXhsValt7c%253D&md5=d301c4721ed676d3fe66d8524ee6e8d1Carbon Monoxide Gas Diffusion Electrolysis that Produces Concentrated C2 Products with High Single-Pass ConversionRipatti, Donald S.; Veltman, Thomas R.; Kanan, Matthew W.Joule (2019), 3 (1), 240-256CODEN: JOULBR; ISSN:2542-4351. (Cell Press)Electrochem. CO conversion is crit. for the development of alternative fuel and chem. syntheses. To be efficient, electrosynthesis must make concd. product streams at high rates with modest potentials, but the combination of these features has not been established for CO or the related CO2 electrolysis. Here we investigate CO electrolysis with gas diffusion electrodes (GDEs) supplied by interdigitated flow fields in electrochem. cells with different ion transport properties. By optimizing gas and ion transport, we show that it is possible to simultaneously achieve high c.d., high selectivity, and high single-pass conversion at moderate cell potentials. Using a cell with the GDE directly contacting a Nafion membrane, we demonstrate >100 mA cm-2 CO redn. to C2 products and direct prodn. of 1.1 M acetate at a cell potential of 2.4 V over 24 h. Our results reveal crit. design features for maximizing the efficiency of C2 electrosynthesis.
- 41Dinh, C. T.; Burdyny, T.; Kibria, M. G.; Seifitokaldani, A.; Gabardo, C. M.; de Arquer, F. P. G.; Kiani, A.; Edwards, J. P.; De Luna, P.; Bushuyev, O. S.; Zou, C.; Quintero-Bermudez, R.; Pang, Y.; Sinton, D.; Sargent, E. H. CO2 Electroreduction to Ethylene via Hydroxide-Mediated Copper Catalysis at an Abrupt Interface. Science 2018, 360 (6390), 783– 787, DOI: 10.1126/science.aas9100[Crossref], [PubMed], [CAS], Google Scholar41https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC1cXpsVCgsL0%253D&md5=0beec1cdcc8939b3eb057cb6b26742f6Carbon dioxide electroreduction to ethylene via hydroxide-mediated copper catalysis at an abrupt interfaceDinh, Cao-Thang; Burdyny, Thomas; Kibria, Md Golam; Seifitokaldani, Ali; Gabardo, Christine M.; Garcia de Arquer, F. Pelayo; Kiani, Amirreza; Edwards, Jonathan P.; De Luna, Phil; Bushuyev, Oleksandr S.; Zou, Chengqin; Quintero-Bermudez, Rafael; Pang, Yuanjie; Sinton, David; Sargent, Edward H.Science (Washington, DC, United States) (2018), 360 (6390), 783-787CODEN: SCIEAS; ISSN:0036-8075. (American Association for the Advancement of Science)Carbon dioxide (CO2) electroredn. could provide a useful source of ethylene, but low conversion efficiency, low prodn. rates, and low catalyst stability limit current systems. Here we report that a copper electrocatalyst at an abrupt reaction interface in an alk. electrolyte reduces CO2 to ethylene with 70% faradaic efficiency at a potential of -0.55 V vs. a reversible hydrogen electrode (RHE). Hydroxide ions on or near the copper surface lower the CO2 redn. and carbon monoxide (CO)-CO coupling activation energy barriers; as a result, onset of ethylene evolution at -0.165 V vs. an RHE in 10 M potassium hydroxide occurs almost simultaneously with CO prodn. Operational stability was enhanced via the introduction of a polymer-based gas diffusion layer that sandwiches the reaction interface between sep. hydrophobic and conductive supports, providing const. ethylene selectivity for an initial 150 operating hours.
- 42Li, Y. C.; Lee, G.; Yuan, T.; Wang, Y.; Nam, D.-H.; Wang, Z.; Garcia de Arquer, F. P.; Lum, Y.; Dinh, C. T.; Voznyy, O.; Sargent, E. H. Co2 Electroreduction From Carbonate Electrolyte. ACS Energy Lett. 2019, 4 (6), 1427– 1431, DOI: 10.1021/acsenergylett.9b00975[ACS Full Text], [CAS], Google Scholar42https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC1MXhtVejtrfF&md5=54aa3a6b76f98e7735a2748b2eb9de57CO2 Electroreduction from Carbonate ElectrolyteLi, Yuguang C.; Lee, Geonhui; Yuan, Tiange; Wang, Ying; Nam, Dae-Hyun; Wang, Ziyun; Garcia de Arquer, F. Pelayo; Lum, Yanwei; Dinh, Cao-Thang; Voznyy, Oleksandr; Sargent, Edward H.ACS Energy Letters (2019), 4 (6), 1427-1431CODEN: AELCCP; ISSN:2380-8195. (American Chemical Society)The process of CO2 valorization-from capture of CO2 to its electrochem. upgrade-requires significant inputs in each of the capture, upgrade, and sepn. steps. Here the authors report an electrolyzer that upgrades carbonate electrolyte from CO2 capture soln. to syngas, achieving 100% C use across the system. A bipolar membrane was used to produce proton in situ to facilitate CO2 release at the membrane:catalyst interface from the carbonate soln. Using a Ag catalyst, the authors generate syngas at a 3:1 H2:CO ratio, and the product is not dild. by CO2 at the gas outlet; the authors generate this pure syngas product stream at a c.d. of 150 mA/cm2 and an energy efficiency of 35%. The carbonate-to-syngas system is stable under a continuous 145 h of catalytic operation. The work demonstrates the benefits of coupling CO2 electrolysis with a CO2 capture electrolyte on the path to practicable CO2 conversion technologies.
- 43Hossain, M. Z.; Rahim, N. A.; Selvaraj, J. A. L. Recent Progress and Development on Power DC-DC Converter Topology, Control, Design and Applications: a Review. Renewable Sustainable Energy Rev. 2018, 81, 205– 230, DOI: 10.1016/j.rser.2017.07.017
Cited By
This article is cited by 14 publications.
- Xin Shen, Maoqing Yao, Ke Sun, Tianshuo Zhao, Yulian He, Chun-Yung Chi, Chongwu Zhou, Paul Daniel Dapkus, Nathan S. Lewis, Shu Hu. Defect-Tolerant TiO2-Coated and Discretized Photoanodes for >600 h of Stable Photoelectrochemical Water Oxidation. ACS Energy Letters 2020, Article ASAP.
- P. Agbo. J–V Decoupling: Independent Control over Current and Potential in Electrocatalysis. The Journal of Physical Chemistry C 2020, Article ASAP.
- Jie He, Csaba Janáky. Recent Advances in Solar-Driven Carbon Dioxide Conversion: Expectations versus Reality. ACS Energy Letters 2020, 5
(6)
, 1996-2014. https://doi.org/10.1021/acsenergylett.0c00645
- Chanon Pornrungroj, Virgil Andrei, Motiar Rahaman, Chawit Uswachoke, Hannah J. Joyce, Dominic S. Wright, Erwin Reisner. Bifunctional Perovskite‐BiVO
4
Tandem Devices for Uninterrupted Solar and Electrocatalytic Water Splitting Cycles. Advanced Functional Materials 2020, 48 , 2008182. https://doi.org/10.1002/adfm.202008182
- Ibadillah A. Digdaya, Ian Sullivan, Meng Lin, Lihao Han, Wen-Hui Cheng, Harry A. Atwater, Chengxiang Xiang. A direct coupled electrochemical system for capture and conversion of CO2 from oceanwater. Nature Communications 2020, 11
(1)
https://doi.org/10.1038/s41467-020-18232-y
- Joshua A. Rabinowitz, Matthew W. Kanan. The future of low-temperature carbon dioxide electrolysis depends on solving one basic problem. Nature Communications 2020, 11
(1)
https://doi.org/10.1038/s41467-020-19135-8
- Da Wang, Yinning He, Na Zhong, Zhiqiao He, Yi Shen, Tao Zeng, Xiaohui Lu, Jun Ma, Shuang Song. In situ chloride-mediated synthesis of TiO2 thin film photoanode with enhanced photoelectrochemical activity for carbamazepine oxidation coupled with simultaneous cathodic H2 production and CO2 conversion to fuels. Journal of Hazardous Materials 2020, , 124563. https://doi.org/10.1016/j.jhazmat.2020.124563
- Charles E. Creissen, Marc Fontecave. Solar‐Driven Electrochemical CO
2
Reduction with Heterogeneous Catalysts. Advanced Energy Materials 2020, 11 , 2002652. https://doi.org/10.1002/aenm.202002652
- Bin Wu, Xu-dong Wang, Xiao-fei Gao, Hao Li, Shan Xi Tian. Dissociative electron attachment to carbon dioxide. Chinese Journal of Chemical Physics 2020, 33
(5)
, 521-531. https://doi.org/10.1063/1674-0068/cjcp2008152
- Sheng Chu, Pengfei Ou, Roksana Tonny Rashid, Pegah Ghamari, Renjie Wang, Hong Nhung Tran, Songrui Zhao, Huiyan Zhang, Jun Song, Zetian Mi. Decoupling Strategy for Enhanced Syngas Generation from Photoelectrochemical CO2 Reduction. iScience 2020, 23
(8)
, 101390. https://doi.org/10.1016/j.isci.2020.101390
- Weilai Yu, Harold J. Fu, Thomas Mueller, Bruce S. Brunschwig, Nathan S. Lewis. Atomic force microscopy: Emerging illuminated and
operando
techniques for solar fuel research. The Journal of Chemical Physics 2020, 153
(2)
, 020902. https://doi.org/10.1063/5.0009858
- Chenyu Xu, Jianan Hong, Pengfei Sui, Mengnan Zhu, Yanwei Zhang, Jing-Li Luo. Standalone Solar Carbon-Based Fuel Production Based on Semiconductors. Cell Reports Physical Science 2020, 1
(7)
, 100101. https://doi.org/10.1016/j.xcrp.2020.100101
- Ya Liu, Liejin Guo. On factors limiting the performance of photoelectrochemical CO
2
reduction. The Journal of Chemical Physics 2020, 152
(10)
, 100901. https://doi.org/10.1063/1.5141390
- Yasuhiko Takeda, Takeshi Morikawa, Naohiko Kato. . Journal of Applied Physics 2020,,, 204503. https://doi.org/10.1063/5.0006310
Abstract
Figure 1
Figure 1. Gas diffusion electrode with Ag-NP catalyst. (a) Cell configuration composed of (1) NiOx or Pt anode, (2) Ag-NPs on Sigracet 29BC carbon paper cathode, (3) anion exchange membrane, (4) CO2 gas inlet and CO/CO2 outlet, (5) acrylic backplate, (6) catholyte chamber, (7) anolyte chamber, (8) reference electrode, (9) GDE (cathode) power connector, and (10) anode power connector. Black arrows indicate the gas flow, and white arrows indicate the electrolyte flow. Note that the backplate (5) is designed to use an interdigitated wire electrode flow field to enhance the interaction between gas and catalysts and improve CO2 utilization (see also Figure S1). (b) Scanning electron microscopy images of carbon paper without (top) and with (bottom) Ag-NP catalyst, secondary electrons image (left row) backscattered electrons image (right row). (c) Illustration of the reverse-assembled GDE cathode cross-section with wetted catalyst and operation for CO2 reduction.
Figure 2
Figure 2. Dark catalysis three-electrode measurement of Ag-NPs GDE. Faradaic efficiency versus GDE potential operated in 1 M KHCO3 (left half of graph) or 1 M KOH (right half of graph) of (a) the reserve-assembled Ag-NP GDE and (b) a standard-assembled Ag-NP GDE. (c) Overpotential versus CO partial current of Ag-NPs GDE for CO2 reduction to CO. Overpotential = |UGDE,RHE + 0.11 V|, JCO ≡ JGDE × fFE,CO. (d) Stability of reserve-assembled and standard-assembled Ag-NPs GDE operated at −0.6 V vs RHE in 1 M KOH.
Figure 3
Figure 3. Light driven PV-GDE measurement (APV = AGDE = 0.31 cm2). (a) Illustration of wire connection between the triple-junction cell and GDE cell. (b) J–U characteristic of Ni anode, solar cell with Ni anode, and Ag-NP gas diffusion cathode under 1 Sun. (c) Current, GDE potential vs RHE, and cell voltage measurement over 20 h duration. (d) Corresponding CO Faradaic efficiency and solar-to-fuel efficiency over the same 20 h duration.
Figure 4
Figure 4. Outdoor assessments of solar-driven PV-GDE in Pasadena, CA (APV = AGDE = 0.31 cm2). The solar irradiance was monitored with a calibrated silicon photodiode. Operating current density J (= JGDE = JPV), cell voltage Ucell, GDE potential UGDE vs RHE, CO Faradaic efficiency fFE,CO, and solar-to-fuel efficiency ηSTF were recorded for a 24 h day cycle.
References
ARTICLE SECTIONSThis article references 43 other publications.
- 1Davis, S. J.; Lewis, N. S.; Shaner, M. R.; Aggarwal, S.; Arent, D.; Azevedo, I. L.; Benson, S. M.; Bradley, T.; Brouwer, J.; Chiang, Y.-M.; Clack, C. T. M.; Cohen, A.; Doig, S.; Edmonds, J.; Fennell, P.; Field, C. B.; Hannegan, B.; Hodge, B.-M.; Hoffert, M. I.; Ingersoll, E.; Jaramillo, P.; Lackner, K. S.; Mach, K. J.; Mastrandrea, M.; Ogden, J.; Peterson, P. F.; Sanchez, D. L.; Sperling, D.; Stagner, J.; Trancik, J. E.; Yang, C.-J.; Caldeira, K. Net-Zero Emissions Energy Systems. Science 2018, 360 (6396), eaas9793 DOI: 10.1126/science.aas9793
- 2Armstrong, R. C.; Wolfram, C.; de Jong, K. P.; Gross, R.; Lewis, N. S.; Boardman, B.; Ragauskas, A. J.; Ehrhardt-Martinez, K.; Crabtree, G.; Ramana, M. V. The Frontiers of Energy. Nature Energy 2016, 1 (1), 15020, DOI: 10.1038/nenergy.2015.20
- 3Shaner, M. R.; Davis, S. J.; Lewis, N. S.; Caldeira, K. Geophysical Constraints on the Reliability of Solar and Wind Power in the United States. Energy Environ. Sci. 2018, 11 (4), 914– 925, DOI: 10.1039/C7EE03029K
- 4Lewis, N. S. Toward Cost-Effective Solar Energy Use. Science 2007, 315 (5813), 798– 801, DOI: 10.1126/science.1137014[Crossref], [PubMed], [CAS], Google Scholar4https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BD2sXhsVShsrs%253D&md5=cc048d85ee8244f9d0d58802cc124395Toward Cost-Effective Solar Energy UseLewis, Nathan S.Science (Washington, DC, United States) (2007), 315 (5813), 798-801CODEN: SCIEAS; ISSN:0036-8075. (American Association for the Advancement of Science)A review. At present, solar energy conversion technologies face cost and scalability hurdles in the technologies required for a complete energy system. To provide a truly widespread primary energy source, solar energy must be captured, converted, and stored in a cost-effective fashion. New developments in nanotechnol., biotechnol., and the materials and phys. sciences may enable step-change approaches to cost-effective, globally scalable systems for solar energy use.
- 5Gray, E. M.; Webb, C. J.; Andrews, J.; Shabani, B.; Tsai, P. J.; Chan, S. L. I. Hydrogen Storage for Off-Grid Power Supply. Int. J. Hydrogen Energy 2011, 36 (1), 654– 663, DOI: 10.1016/j.ijhydene.2010.09.051[Crossref], [CAS], Google Scholar5https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC3MXht1Gjt7w%253D&md5=39d2a80bd405461715d573f44da5ea62Hydrogen storage for off-grid power supplyGray, E. MacA.; Webb, C. J.; Andrews, J.; Shabani, B.; Tsai, P. J.; Chan, S. L. I.International Journal of Hydrogen Energy (2011), 36 (1), 654-663CODEN: IJHEDX; ISSN:0360-3199. (Elsevier Ltd.)The use of intermittent renewable energy sources for power supply to off-grid electricity consumers depends on energy storage technol. to guarantee continuous supply. Potential applications of storage-guaranteed systems range from small installations for remote telecoms, water-pumping and single dwellings, to farms and whole communities for whom grid connection is too expensive or otherwise infeasible, to industrial, military and humanitarian uses. In this paper we explore some of the tech. issues surrounding the use of hydrogen storage, in conjunction with a PEM electrolyzer and PEM fuel cell, to guarantee electricity supply when the energy source is intermittent, most typically solar photovoltaic. We advocate metal-hydride storage and compare its energy d. to that of Li-ion battery storage, concluding that a significantly smaller package is possible with metal-hydride storage. A simple approach to match the output of a photovoltaic array to an electrolyzer is presented. The properties required for the metal-hydride storage material to interface the electrolyzer to the fuel cell are discussed in detail. Relatively conventional Mischmetal-based AB5 alloys are suitable for this application.
- 6Jia, J.; Seitz, L. C.; Benck, J. D.; Huo, Y.; Chen, Y.; Ng, J. W. D.; Bilir, T.; Harris, J. S.; Jaramillo, T. F. Solar Water Splitting by Photovoltaic-Electrolysis with a Solar-to-Hydrogen Efficiency Over 30%. Nat. Commun. 2016, 7, 13237, DOI: 10.1038/ncomms13237[Crossref], [PubMed], [CAS], Google Scholar6https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC28XhvVSisLfI&md5=2e1f9254a9aa1862b6a03635a0429949Solar water splitting by photovoltaic-electrolysis with a solar-to-hydrogen efficiency over 30%Jia, Jieyang; Seitz, Linsey C.; Benck, Jesse D.; Huo, Yijie; Chen, Yusi; Ng, Jia Wei Desmond; Bilir, Taner; Harris, James S.; Jaramillo, Thomas F.Nature Communications (2016), 7 (), 13237CODEN: NCAOBW; ISSN:2041-1723. (Nature Publishing Group)Hydrogen prodn. via electrochem. water splitting is a promising approach for storing solar energy. For this technol. to be economically competitive, it is crit. to develop water splitting systems with high solar-to-hydrogen (STH) efficiencies. Here we report a photovoltaic-electrolysis system with the highest STH efficiency for any water splitting technol. to date, to the best of our knowledge. Our system consists of two polymer electrolyte membrane electrolyzers in series with one InGaP/GaAs/GaInNAsSb triple-junction solar cell, which produces a large-enough voltage to drive both electrolyzers with no addnl. energy input. The solar concn. is adjusted such that the max. power point of the photovoltaic is well matched to the operating capacity of the electrolyzers to optimize the system efficiency. The system achieves a 48-h av. STH efficiency of 30%. These results demonstrate the potential of photovoltaic-electrolysis systems for cost-effective solar energy storage.
- 7Young, J. L.; Steiner, M. A.; Döscher, H.; France, R. M.; Turner, J. A.; Deutsch, T. G. Direct Solar-to-Hydrogen Conversion via Inverted Metamorphic Multi-Junction Semiconductor Architectures. Nature Energy 2017, 2, 17028, DOI: 10.1038/nenergy.2017.28[Crossref], [CAS], Google Scholar7https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC2sXot1ykuro%253D&md5=04ac8805e7de2455968709947216ebbcDirect solar-to-hydrogen conversion via inverted metamorphic multi-junction semiconductor architecturesYoung, James L.; Steiner, Myles A.; Doscher, Henning; France, Ryan M.; Turner, John A.; Deutsch, Todd G.Nature Energy (2017), 2 (4), 17028CODEN: NEANFD; ISSN:2058-7546. (Nature Publishing Group)Solar water splitting via multi-junction semiconductor photoelectrochem. cells provides direct conversion of solar energy to stored chem. energy as hydrogen bonds. Economical hydrogen prodn. demands high conversion efficiency to reduce balance-of-systems costs. For sufficient photovoltage, water-splitting efficiency is proportional to the device photocurrent, which can be tuned by judicious selection and integration of optimal semiconductor bandgaps. Here, we demonstrate highly efficient, immersed water-splitting electrodes enabled by inverted metamorphic epitaxy and a transparent graded buffer that allows the bandgap of each junction to be independently varied. Voltage losses at the electrolyte interface are reduced by 0.55 V over traditional, uniformly p-doped photocathodes by using a buried p-n junction. Advanced on-sun benchmarking, spectrally cor. and validated with incident photon-to-current efficiency, yields over 16% solar-to-hydrogen efficiency with GaInP/GaInAs tandem absorbers, representing a 60% improvement over the classical, high-efficiency tandem III-V device.
- 8Cheng, W.-H.; Richter, M. H.; May, M. M.; Ohlmann, J.; Lackner, D.; Dimroth, F.; Hannappel, T.; Atwater, H. A.; Lewerenz, H. J. Monolithic Photoelectrochemical Device for Direct Water Splitting with 19% Efficiency. ACS Energy Lett. 2018, 3 (8), 1795– 1800, DOI: 10.1021/acsenergylett.8b00920[ACS Full Text], [CAS], Google Scholar8https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC1cXhtFyqtbrK&md5=0597725a2cef1271ca423c4764a23c9cMonolithic Photoelectrochemical Device for Direct Water Splitting with 19% EfficiencyCheng, Wen-Hui; Richter, Matthias H.; May, Matthias M.; Ohlmann, Jens; Lackner, David; Dimroth, Frank; Hannappel, Thomas; Atwater, Harry A.; Lewerenz, Hans-JoachimACS Energy Letters (2018), 3 (8), 1795-1800CODEN: AELCCP; ISSN:2380-8195. (American Chemical Society)Efficient unassisted solar water splitting, a pathway to storable renewable energy in the form of chem. bonds, requires optimization of a photoelectrochem. device based on photovoltaic tandem heterojunctions. We report a monolithic photocathode device architecture that exhibits significantly reduced surface reflectivity, minimizing parasitic light absorption and reflection losses. A tailored multifunctional cryst. titania interphase layer acts as a corrosion protection layer, with favorable band alignment between the semiconductor conduction band and the energy level for water redn., facilitating electron transport at the cathode-electrolyte interface. It also provides a favorable substrate for adhesion of high-activity Rh catalyst nanoparticles. Under simulated AM 1.5G irradn., solar-to-H efficiencies of 19.3 and 18.5% are obtained in acidic and neutral electrolytes, resp. The system reaches 0.85 of the theor. limit for photoelectrochem. water splitting for the energy gap combination employed in the tandem-junction photoelectrode structure.
- 9Seh, Z. W.; Kibsgaard, J.; Dickens, C. F.; Chorkendorff, I.; Nørskov, J. K.; Jaramillo, T. F. Combining Theory and Experiment in Electrocatalysis: Insights Into Materials Design. Science 2017, 355 (6321), eaad4998 DOI: 10.1126/science.aad4998
- 10Schreier, M.; Héroguel, F.; Steier, L.; Ahmad, S.; Luterbacher, J. S.; Mayer, M. T.; Luo, J.; Grätzel, M. Solar Conversion of CO2 To CO Using Earth-Abundant Electrocatalysts Prepared by Atomic Layer Modification of CuO. Nature Energy 2017, 2 (7), 17087, DOI: 10.1038/nenergy.2017.87[Crossref], [CAS], Google Scholar10https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC1cXitVeht7g%253D&md5=4efbebcfad2748d46a1361c22c435248Solar conversion of CO2 to CO using Earth-abundant electrocatalysts prepared by atomic layer modification of CuOSchreier, Marcel; Heroguel, Florent; Steier, Ludmilla; Ahmad, Shahzada; Luterbacher, Jeremy S.; Mayer, Matthew T.; Luo, Jingshan; Gratzel, MichaelNature Energy (2017), 2 (7), 17087CODEN: NEANFD; ISSN:2058-7546. (Nature Research)The solar-driven electrochem. redn. of CO2 to fuels and chems. provides a promising way for closing the anthropogenic carbon cycle. However, the lack of selective and Earth-abundant catalysts able to achieve the desired transformation reactions in an aq. matrix presents a substantial impediment as of today. Here we introduce at. layer deposition of SnO2 on CuO nanowires as a means for changing the wide product distribution of CuO-derived CO2 redn. electrocatalysts to yield predominantly CO. The activity of this catalyst towards oxygen evolution enables us to use it both as the cathode and anode for complete CO2 electrolysis. In the resulting device, the electrodes are sepd. by a bipolar membrane, allowing each half-reaction to run in its optimal electrolyte environment. Using a GaInP/GaInAs/Ge photovoltaic we achieve the solar-driven splitting of CO2 into CO and oxygen with a bifunctional, sustainable and all Earth-abundant system at an efficiency of 13.4%.
- 11Gurudayal; Beeman, J. W.; Bullock, J.; Wang, H.; Eichhorn, J.; Towle, C.; Javey, A.; Toma, F. M.; Mathews, N.; Ager, J. W., III Si Photocathode with Ag-Supported Dendritic Cu Catalyst for CO2 Reduction. Energy Environ. Sci. 2019, 12 (3), 1068– 1077, DOI: 10.1039/C8EE03547D[Crossref], [CAS], Google Scholar11https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC1MXjt1eqtbk%253D&md5=d83b4701612dc73fddb583a3e5d1da15Si photocathode with Ag-supported dendritic Cu catalyst for CO2 reductionGurudayal; Beeman, Jeffrey W.; Bullock, James; Wang, Hao; Eichhorn, Johanna; Towle, Clarissa; Javey, Ali; Toma, Francesca M.; Mathews, Nripan; Ager, Joel W.Energy & Environmental Science (2019), 12 (3), 1068-1077CODEN: EESNBY; ISSN:1754-5706. (Royal Society of Chemistry)Si photocathodes integrated with Ag-supported dendritic Cu catalysts are used to perform light-driven redn. of CO2 to C2 and C3 products in aq. soln. A back illumination geometry with an n-type Si absorber was used to permit the use of absorbing metallic catalysts. Selective carrier collection was accomplished by a p+ implantation on the illumination side and an n+ implantation followed by at. layer deposition of TiO2 on the electrolyte site. The Ag-supported dendritic Cu CO2 redn. catalyst was formed by evapn. of Ag followed by high-rate electrodeposition of Cu to form a high surface area structure. Under simulated 1 sun illumination in 0.1 M CsHCO3 satd. with CO2, the photovoltage generated by the Si (∼600 mV) enables C2 and C3 products to be produced at -0.4 vs. Texturing of both sides of the Si increases the light-limited c.d., due to reduced reflection on the illumination side, and also deceases the onset potential. Under simulated diurnal illumination conditions photocathodes maintain over 60% faradaic efficiency to hydrocarbon and oxygenate products (mainly ethylene, ethanol, propanol) for several days. After 10 days of testing, contamination from the counter electrode is obsd., which causes an increase in hydrogen prodn. This effect is mitigated by a regeneration procedure which restores the original catalyst selectivity. A tandem, self-powered CO2 redn. device was formed by coupling a Si photocathode with two series-connected semitransparent CH3NH3PbI3 perovskite solar cells, achieving an efficiency for the conversion of sunlight to hydrocarbons and oxygenates of 1.5% (3.5% for all products).
- 12Zhou, X.; Liu, R.; Sun, K.; Chen, Y.; Verlage, E.; Francis, S. A.; Lewis, N. S.; Xiang, C. Solar-Driven Reduction of 1 atm of CO2 To Formate at 10% Energy-Conversion Efficiency by Use of a TiO2-Protected III–V Tandem Photoanode in Conjunction with a Bipolar Membrane and a Pd/C Cathode. ACS Energy Lett. 2016, 1 (4), 764– 770, DOI: 10.1021/acsenergylett.6b00317[ACS Full Text], [CAS], Google Scholar12https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC28XhsV2hs77J&md5=ce1fce3fa43e3a3711ed47debe205586Solar-Driven Reduction of 1 atm of CO2 to Formate at 10% Energy-Conversion Efficiency by Use of a TiO2-Protected III-V Tandem Photoanode in Conjunction with a Bipolar Membrane and a Pd/C CathodeZhou, Xinghao; Liu, Rui; Sun, Ke; Chen, Yikai; Verlage, Erik; Francis, Sonja A.; Lewis, Nathan S.; Xiang, ChengxiangACS Energy Letters (2016), 1 (4), 764-770CODEN: AELCCP; ISSN:2380-8195. (American Chemical Society)A solar-driven CO2 redn. (CO2R) cell was constructed, consisting of a tandem GaAs/InGaP/TiO2/Ni photoanode in 1.0M KOH(aq) (pH = 13.7) to facilitate the oxygen-evolution reaction (OER), a Pd/C nanoparticle-coated Ti mesh cathode in 2.8M KHCO3(aq) (pH = 8.0) to perform the CO2R reaction, and a bipolar membrane to allow for steady-state operation of the catholyte and anolyte at different bulk pH values. At the operational c.d. of 8.5 mA cm-2, in 2.8M KHCO3(aq), the cathode exhibited <100 mV overpotential and >94% faradaic efficiency for the redn. of 1 atm of CO2(g) to formate. The anode exhibited a 320 ± 7 mV overpotential for the OER in 1.0M KOH(aq), and the bipolar membrane exhibited ∼480 mV voltage loss with minimal product crossovers and >90 and >95% selectivity for protons and hydroxide ions, resp. The bipolar membrane facilitated coupling between two electrodes and electrolytes, one for the CO2R reaction and one for the OER, that typically operate at mutually different pH values and produced a lower total cell overvoltage than known single-electrolyte CO2R systems while exhibiting ∼10% solar-to-fuels energy-conversion efficiency.
- 13Romero Cuellar, N. S.; Wiesner-Fleischer, K.; Fleischer, M.; Rucki, A.; Hinrichsen, O. Advantages of CO Over CO2 As Reactant for Electrochemical Reduction to Ethylene, Ethanol and N-Propanol on Gas Diffusion Electrodes at High Current Densities. Electrochim. Acta 2019, 307, 164– 175, DOI: 10.1016/j.electacta.2019.03.142[Crossref], [CAS], Google Scholar13https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC1MXmslOnu70%253D&md5=93b32c2a5974c6d255d14802330985f4Advantages of CO over CO2 as reactant for electrochemical reduction to ethylene, ethanol and n-propanol on gas diffusion electrodes at high current densitiesRomero Cuellar, N. S.; Wiesner-Fleischer, K.; Fleischer, M.; Rucki, A.; Hinrichsen, O.Electrochimica Acta (2019), 307 (), 164-175CODEN: ELCAAV; ISSN:0013-4686. (Elsevier Ltd.)The electrochem. conversion of CO2 to value-added chems. is a technol. gaining broader interest as society moves towards a carbon-neutral circular economy. Nonetheless, there are still several challenges to overcome before this technol. can be applied as an industrial process. In the reaction path of the electrochem. redn. of CO2 with Cu as an electrocatalyst, it is known that carbon monoxide is the key intermediate to chems. such as ethylene, ethanol, and n-propanol. However, a better understanding of the electrochem. redn. of CO is still necessary to improve selectivity and efficiency at high current densities. In this work, the electrochem. redn. of CO2 and CO towards C2 and C3 products is investigated using gas diffusion electrodes in a flow cell. Thereby the electrochem. reaction is not limited by the soly. of the feed gas in the electrolyte, and current densities of industrial relevance can be achieved. The electrodes are prepd. using com. Cu-powders consisting either of nano- or microparticles that are deposited on gas diffusion layers. Potentiostatic expts. show that with CO as the reactant, higher current densities for C2 and C3 products can be achieved at lower working electrode potentials compared to CO2 as the reactant. Galvanostatic CO electrochem. redn. at -300 mA cm-2 with Cu-nanoparticles (40-60 nm) results in a cumulative Faradaic efficiency of 89% for C2 and C3 products. This represents a two-fold increase in selectivity to ethylene and a three-fold increase towards ethanol and n-propanol compared to the selectivity obtained with CO2 as the reactant. This enhancement of selectivity for C2 and C3 products at current densities of industrial relevance with CO as reactant provides a new perspective regarding a two-step electrochem. redn. of CO2.
- 14Zhou, X.; Xiang, C. Comparative Analysis of Solar-to-Fuel Conversion Efficiency: a Direct, One-Step Electrochemical CO2 Reduction Reactor Versus a Two-Step, Cascade Electrochemical CO2 Reduction Reactor. ACS Energy Lett. 2018, 3 (8), 1892– 1897, DOI: 10.1021/acsenergylett.8b01077[ACS Full Text], [CAS], Google Scholar14https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC1cXhtlahu7rE&md5=b6d344c1806c6c31ad405ca45c6fc210Comparative Analysis of Solar-to-Fuel Conversion Efficiency: A Direct, One-Step Electrochemical CO2 Reduction Reactor versus a Two-Step, Cascade Electrochemical CO2 Reduction ReactorZhou, Xinghao; Xiang, ChengxiangACS Energy Letters (2018), 3 (8), 1892-1897CODEN: AELCCP; ISSN:2380-8195. (American Chemical Society)The solar-to-fuel conversion efficiencies of a direct, one-step reactor that electrochem. reduces CO2 to C2H6O and a two-step, cascade reactor that electrochem. reduces CO2 CO followed by a subsequent electrochem. redn. of CO to C2H6O were evaluated and compared quant. By leveraging the efficient and selective first two-electron, two proton process from CO2 to CO, the optimal solar-to-fuel conversion efficiency of the two-step reactor was higher than that of the one-step reactor at all cathodic overpotential and Faradaic efficiency combinations. The anal. shows that in some electrocatalyst performance regions with high cathodic overpotentials a relative improvement in the solar-to-fuel conversion efficiency as high as 54% can be obtained by using the two-step reactor. The alternative, two-step CO2 reactor design can provide new pathways to efficient and selective CO2 redn. to higher redn. products.
- 15Delacourt, C.; Ridgway, P. L.; Kerr, J. B.; Newman, J. Design of an Electrochemical Cell Making Syngas (CO + H2) From CO2 And H2O Reduction at Room Temperature. J. Electrochem. Soc. 2008, 155 (1), B42– B49, DOI: 10.1149/1.2801871
- 16Schulz, H. Short History and Present Trends of Fischer–Tropsch Synthesis. Appl. Catal., A 1999, 186 (1–2), 3– 12, DOI: 10.1016/S0926-860X(99)00160-X[Crossref], [CAS], Google Scholar16https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADyaK1MXmtVOkurY%253D&md5=01ee90af12a4d3a00fc0b39cbd26317bShort history and present trends of Fischer-Tropsch synthesisSchulz, H.Applied Catalysis, A: General (1999), 186 (1,2), 3-12CODEN: ACAGE4; ISSN:0926-860X. (Elsevier Science B.V.)A review, with 68 refs. Due to the large vol. of existing literature on Fischer-Tropsch (FT) synthesis, the diversity of the subject and the actually reoriented interest, it seemed indicated to write a historical sketch about the process, putting also emphasis on present trends and future options. Thus history and trends have been divided into several lines which are elaborated individually. Of course, presenting history and trends of FT synthesis on a few pages means generalizing from many individual investigations and developments and also selection of only a few citations. So I want to apologize for all the contributions to science and technol. around FT synthesis which I have missed to include into the article.
- 17Nielsen, D. U.; Hu, X.-M.; Daasbjerg, K.; Skrydstrup, T. Chemically and Electrochemically Catalysed Conversion of CO2 To CO with Follow-Up Utilization to Value-Added Chemicals. Nature Catalysis 2018, 1 (4), 244– 254, DOI: 10.1038/s41929-018-0051-3[Crossref], [CAS], Google Scholar17https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC1MXpvVyrtbc%253D&md5=beeabaa45d07b5cfb0d698e643c88949Chemically and electrochemically catalysed conversion of CO2 to CO with follow-up utilization to value-added chemicalsNielsen, Dennis U.; Hu, Xin-Ming; Daasbjerg, Kim; Skrydstrup, TroelsNature Catalysis (2018), 1 (4), 244-254CODEN: NCAACP; ISSN:2520-1158. (Nature Research)A review. Carbon dioxide is ubiquitous and a vital mol. for maintaining life on our planet. However, the ever-increasing emission of anthropogenic CO2 into our atm. has provoked dramatic climate changes. In principle, CO2 could represent an important one-carbon building block for the chem. industry, yet its high thermodn. and kinetic stability has limited its applicability to only a handful of industrial applications. On the other hand, carbon monoxide represents a more versatile reagent applied in many industrial transformations. Here we review the different methods for converting CO2 to CO with specific focus on the reverse water gas shift reaction, main element reductants, and electrochem. protocols applying homogeneous and heterogeneous catalysts. Particular emphasis is given to synthetic methods that couple the deoxygenation step with a follow-up carbonylation step for the synthesis of carbonyl-contg. mols., thus avoiding the need to handle or store this toxic but highly synthetically useful diat. gas.
- 18Lum, Y.; Ager, J. W., III. Sequential Catalysis Controls Selectivity in Electrochemical CO2 Reduction on Cu. Energy Environ. Sci. 2018, 11 (10), 2935– 2944, DOI: 10.1039/C8EE01501E[Crossref], [CAS], Google Scholar18https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC1cXht1yiu7nM&md5=d3276f3a384ae3fb6760eb13d42ba6adSequential catalysis controls selectivity in electrochemical CO2 reduction on CuLum, Yanwei; Ager, Joel W.Energy & Environmental Science (2018), 11 (10), 2935-2944CODEN: EESNBY; ISSN:1754-5706. (Royal Society of Chemistry)Electrochem. redn. of CO2 in aq. media is a strategy for sustainable prodn. of fuels and commodity chems. Cu is the only catalyst which converts CO2 to significant quantities of hydrocarbons and oxygenates. Here we demonstrate that oxygenate products can be favored over hydrocarbons by positioning a local source of CO generated by a CO producing catalyst (Au or Ag) in close proximity to a Cu catalyst. Use of a bimetallic device comprising interdigitated and independently controllable lines of Au and Cu allows the local CO concn. to be modulated. Notably, diffusional simulations show that the satn. concn. of CO can be exceeded locally. Actuating both the Au and Cu lines increases the oxygenate to ethylene ratio compared to actuating Cu only. Increasing the relative area of CO-producing Au relative to Cu also increases this ratio. These insights are translated into a second bimetallic system comprising Cu dots/lines patterned directly onto a Ag substrate, allowing for the distance between Cu and the CO generating metal to be precisely controlled. Controlling the relative areas of Ag and Cu allows for tuning of the oxygenate to ethylene ratio from 0.59 to 2.39 and an increase in oxygenate faradaic efficiency from 21.4% to 41.4%, while maintaining the selectivity to C2/C3 products in the 50-65% range. We attribute this change in selectivity to be due to an increased *CO coverage on Cu. By utilizing diffusional transport of CO to the Cu, a sequential catalysis pathway is created which allows for the control of oxygenate selectivity in aq. CO2 redn.
- 19Jouny, M.; Luc, W.; Jiao, F. High-Rate Electroreduction of Carbon Monoxide to Multi-Carbon Products. Nature Catalysis 2018, 1 (10), 748– 755, DOI: 10.1038/s41929-018-0133-2[Crossref], [CAS], Google Scholar19https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC1MXhtFGisL%252FK&md5=d0e97e6e1ae97bbde8a7c4b473693abcHigh-rate electroreduction of carbon monoxide to multi-carbon productsJouny, Matthew; Luc, Wesley; Jiao, FengNature Catalysis (2018), 1 (10), 748-755CODEN: NCAACP; ISSN:2520-1158. (Nature Research)Carbon monoxide electrolysis has previously been reported to yield enhanced multi-carbon (C2+) Faradaic efficiencies of up to ∼55%, but only at low reaction rates. This is due to the low soly. of CO in aq. electrolytes and operation in batch-type reactors. Here, we present a high-performance CO flow electrolyzer with a well controlled electrode-electrolyte interface that can reach total current densities of up to 1 A cm-2, together with improved C2+ selectivities. Computational transport modeling and isotopic C18O redn. expts. suggest that the enhanced activity is due to a higher surface pH under CO redn. conditions, which facilitates the prodn. of acetate. At optimal operating conditions, we achieve a C2+ Faradaic efficiency of ∼91% with a C2+ partial c.d. over 630 mA cm-2. Further investigations show that maintaining an efficient triple-phase boundary at the electrode-electrolyte interface is the most crit. challenge in achieving a stable CO/CO2 electrolysis process at high rates.
- 20Welch, A. J.; DuChene, J. S.; Tagliabue, G.; Davoyan, A.; Cheng, W.-H.; Atwater, H. A. Nanoporous Gold as a Highly Selective and Active Carbon Dioxide Reduction Catalyst. ACS Appl. Energy Mater. 2019, 2 (1), 164– 170, DOI: 10.1021/acsaem.8b01570[ACS Full Text], [CAS], Google Scholar20https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC1cXisF2lurzM&md5=1c0bba179a63dc7ac0d3af6d2bd1eb5fNanoporous Gold as a Highly Selective and Active Carbon Dioxide Reduction CatalystWelch, Alex J.; DuChene, Joseph S.; Tagliabue, Giulia; Davoyan, Artur; Cheng, Wen-Hui; Atwater, Harry A.ACS Applied Energy Materials (2019), 2 (1), 164-170CODEN: AAEMCQ; ISSN:2574-0962. (American Chemical Society)Electrochem. conversion of CO2 into useful chems. is a promising approach for transforming CO2 into sustainably produced fuels and/or chem. feedstocks for industrial synthesis. The authors report that nanoporous Au (np-Au) films, with pore sizes ranging from 10 to 30 nm, represent promising electrocatalytic architectures for the CO2 redn. reaction (CO2RR) due to their large electrochem. active surface area, relative abundance of grain boundaries, and ability to support pH gradients inside the nanoporous network. Electrochem. studies show that np-Au films support partial current densities for the conversion of CO2 to CO >6 mA cm-2 at a faradaic efficiency of ∼99% in aq. electrolytes (50 mM K2CO3 satd. with CO2). Also, np-Au films are able to maintain faradaic efficiency >80% for CO prodn. over prolonged periods of continuous operation (110 h). Electrocatalytic expts. at different electrolyte concns. demonstrate that the pore diam. of nanoporous cathodes represents a crit. parameter for creating and controlling local pH gradients inside the porous network of metal ligaments. These results demonstrate the merits of nanoporous metal films for the CO2RR and offer an interesting architecture for highly selective electrocatalysis capable of sustaining high catalytic currents over prolonged periods.
- 21Chen, Y.; Li, C. W.; Kanan, M. W. Aqueous CO2 Reduction at Very Low Overpotential on Oxide-Derived Au Nanoparticles. J. Am. Chem. Soc. 2012, 134 (49), 19969– 19972, DOI: 10.1021/ja309317u[ACS Full Text], [CAS], Google Scholar21https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC38Xhslalu7rN&md5=bc3f0624d8d46f9e16aca5d1a0f66420Aqueous CO2 Reduction at Very Low Overpotential on Oxide-Derived Au NanoparticlesChen, Yihong; Li, Christina W.; Kanan, Matthew W.Journal of the American Chemical Society (2012), 134 (49), 19969-19972CODEN: JACSAT; ISSN:0002-7863. (American Chemical Society)Carbon dioxide redn. is an essential component of many prospective technologies for the renewable synthesis of carbon-contg. fuels. Known catalysts for this reaction generally suffer from low energetic efficiency, poor product selectivity, and rapid deactivation. It is shown that the redn. of thick Au oxide films results in the formation of Au nanoparticles (oxide-derived Au) that exhibit highly selective CO2 redn. to CO in water at overpotentials as low as 140 mV and retain their activity for at least 8 h. Under identical conditions, polycryst. Au electrodes and several other nanostructured Au electrodes prepd. via alternative methods require at least 200 mV of addnl. overpotential to attain comparable CO2 redn. activity and rapidly lose their activity. Electrokinetic studies indicate that the improved catalysis is linked to dramatically increased stabilization of the CO2•- intermediate on the surfaces of the oxide-derived Au electrodes.
- 22Hatsukade, T.; Kuhl, K. P.; Cave, E. R.; Abram, D. N.; Jaramillo, T. F. Insights Into the Electrocatalytic Reduction of CO2 On Metallic Silver Surfaces. Phys. Chem. Chem. Phys. 2014, 16 (27), 13814– 13819, DOI: 10.1039/C4CP00692E[Crossref], [PubMed], [CAS], Google Scholar22https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC2cXhtVantrjM&md5=639d007871b7b111fbc30803d23add94Insights into the electrocatalytic reduction of CO2 on metallic silver surfacesHatsukade, Toru; Kuhl, Kendra P.; Cave, Etosha R.; Abram, David N.; Jaramillo, Thomas F.Physical Chemistry Chemical Physics (2014), 16 (27), 13814-13819CODEN: PPCPFQ; ISSN:1463-9076. (Royal Society of Chemistry)The electrochem. redn. of CO2 could allow for a sustainable process by which renewable energy from wind and solar are used directly in the prodn. of fuels and chems. In this work we investigated the potential dependent activity and selectivity of the electrochem. redn. of CO2 on metallic silver surfaces under ambient conditions. Our results deepen our understanding of the surface chem. and provide insight into the factors important to designing better catalysts for the reaction. The high sensitivity of our exptl. methods for identifying and quantifying products of reaction allowed for the observation of six redn. products including CO and hydrogen as major products and formate, methane, methanol, and ethanol as minor products. By quantifying the potential-dependent behavior of all products, we provide insights into kinetics and mechanisms at play, in particular involving the prodn. of hydrocarbons and alcs. on catalysts with weak CO binding energy as well as the formation of a C-C bond required to produce ethanol.
- 23Asadi, M.; Kim, K.; Liu, C.; Addepalli, A. V.; Abbasi, P.; Yasaei, P.; Phillips, P.; Behranginia, A.; Cerrato, J. M.; Haasch, R.; Zapol, P.; Kumar, B.; Klie, R. F.; Abiade, J.; Curtiss, L. A.; Salehi-Khojin, A. Nanostructured Transition Metal Dichalcogenide Electrocatalysts for CO2 Reduction in Ionic Liquid. Science 2016, 353 (6298), 467– 470, DOI: 10.1126/science.aaf4767[Crossref], [PubMed], [CAS], Google Scholar23https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC28Xht1ehtL7M&md5=b2a2c08639c77e7ca8afd6c4c1a08bd4Nanostructured transition metal dichalcogenide electrocatalysts for CO2 reduction in ionic liquidAsadi, Mohammad; Kim, Kibum; Liu, Cong; Addepalli, Aditya Venkata; Abbasi, Pedram; Yasaei, Poya; Phillips, Patrick; Behranginia, Amirhossein; Cerrato, Jose M.; Haasch, Richard; Zapol, Peter; Kumar, Bijandra; Klie, Robert F.; Abiade, Jeremiah; Curtiss, Larry A.; Salehi-Khojin, AminScience (Washington, DC, United States) (2016), 353 (6298), 467-470CODEN: SCIEAS; ISSN:0036-8075. (American Association for the Advancement of Science)Conversion of carbon dioxide (CO2) into fuels is an attractive soln. to many energy and environmental challenges. However, the chem. inertness of CO2 renders many electrochem. and photochem. conversion processes inefficient. A transition metal dichalcogenide nanoarchitecture is reported for catalytic electrochem. CO2 conversion to carbon monoxide (CO) in an ionic liq. It is found that tungsten diselenide nanoflakes show a c.d. of 18.95 mA per square centimeter, CO faradaic efficiency of 24%, and CO formation turnover frequency of 0.28 per s at a low overpotential of 54 mV. The catalyst is also applied in a light-harvesting artificial leaf platform that concurrently oxidized water in the absence of any external potential.
- 24Asadi, M.; Kumar, B.; Behranginia, A.; Rosen, B. A.; Baskin, A.; Repnin, N.; Pisasale, D.; Phillips, P.; Zhu, W.; Haasch, R.; Klie, R. F.; Král, P.; Abiade, J.; Salehi-Khojin, A. Robust Carbon Dioxide Reduction on Molybdenum Disulphide Edges.. Nat. Commun. 2014, 5, 4470, DOI: 10.1038/ncomms5470[Crossref], [PubMed], [CAS], Google Scholar24https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC2cXhvF2lsr%252FF&md5=81f74b1c1a0ee5987742eb6cbfb03c9eRobust carbon dioxide reduction on molybdenum disulphide edgesAsadi, Mohammad; Kumar, Bijandra; Behranginia, Amirhossein; Rosen, Brian A.; Baskin, Artem; Repnin, Nikita; Pisasale, Davide; Phillips, Patrick; Zhu, Wei; Haasch, Richard; Klie, Robert F.; Kral, Petr; Abiade, Jeremiah; Salehi-Khojin, AminNature Communications (2014), 5 (), 4470CODEN: NCAOBW; ISSN:2041-1723. (Nature Publishing Group)Electrochem. redn. of carbon dioxide has been recognized as an efficient way to convert carbon dioxide to energy-rich products. Noble metals (for example, gold and silver) have been demonstrated to reduce carbon dioxide at moderate rates and low overpotentials. Nevertheless, the development of inexpensive systems with an efficient carbon dioxide redn. capability remains a challenge. Here we identify molybdenum disulfide as a promising cost-effective substitute for noble metal catalysts. We uncover that molybdenum disulfide shows superior carbon dioxide redn. performance compared with the noble metals with a high c.d. and low overpotential (54 mV) in an ionic liq. Scanning transmission electron microscopy anal. and first principle modeling reveal that the molybdenum-terminated edges of molybdenum disulfide are mainly responsible for its catalytic performance due to their metallic character and a high d-electron d. This is further exptl. supported by the carbon dioxide redn. performance of vertically aligned molybdenum disulfide.
- 25Cheng, T.; Huang, Y.; Xiao, H.; Goddard, W. A. Predicted Structures of the Active Sites Responsible for the Improved Reduction of Carbon Dioxide by Gold Nanoparticles. J. Phys. Chem. Lett. 2017, 8 (14), 3317– 3320, DOI: 10.1021/acs.jpclett.7b01335[ACS Full Text], [CAS], Google Scholar25https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC2sXhtFSnu7vN&md5=3108377ea91d863955ec4e554eb49807Predicted Structures of the Active Sites Responsible for the Improved Reduction of Carbon Dioxide by Gold NanoparticlesCheng, Tao; Huang, Yufeng; Xiao, Hai; Goddard, William A.Journal of Physical Chemistry Letters (2017), 8 (14), 3317-3320CODEN: JPCLCD; ISSN:1948-7185. (American Chemical Society)Gold (Au) nanoparticles (NPs) are known exptl. to reduce carbon dioxide (CO2) to carbon monoxide (CO), with far superior performance to Au foils. To obtain guidance in designing improved CO2 catalysts, we want to understand the nature of the active sites on Au NPs. Here, we employed multiscale atomistic simulations to computationally synthesize and characterize a 10 nm thick Au NP on a carbon nanotube (CNT) support, and then we located active sites from quantum mechanics (QM) calcns. on 269 randomly selected sites. The std. scaling relation is that the formation energy of *COOH (ΔE*COOH) is proportional to the binding energy of *CO (Ebinding*CO); therefore, decreasing ΔE*COOH to boost the CO2 redn. reaction (CO2RR) causes an increase of Ebinding*CO that retards CO2RR. We show that the NPs have superior CO2RR because there are many sites at the twin boundaries that significantly break this scaling relation.
- 26Singh, M. R.; Kwon, Y.; Lum, Y.; Ager, J. W., III; Bell, A. T. Hydrolysis of Electrolyte Cations Enhances the Electrochemical Reduction of CO2 Over Ag and Cu. J. Am. Chem. Soc. 2016, 138 (39), 13006– 13012, DOI: 10.1021/jacs.6b07612[ACS Full Text], [CAS], Google Scholar26https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC28XhsFWjur3P&md5=e1a7c5a8f8ee0451fe834febe8b1d123Hydrolysis of Electrolyte Cations Enhances the Electrochemical Reduction of CO2 over Ag and CuSingh, Meenesh R.; Kwon, Youngkook; Lum, Yanwei; Ager, Joel W.; Bell, Alexis T.Journal of the American Chemical Society (2016), 138 (39), 13006-13012CODEN: JACSAT; ISSN:0002-7863. (American Chemical Society)Electrolyte cation size is known to influence the electrochem. redn. of CO2 over metals; however, a satisfactory explanation for this phenomenon was not developed. The authors report here that these effects can be attributed to a previously unrecognized consequence of cation hydrolysis occurring in the vicinity of the cathode. With increasing cation size, the pKa for cation hydrolysis decreases and is sufficiently low for hydrated K+, Rb+, and Cs+ to serve as buffering agents. Buffering lowers the pH near the cathode, increasing the local concn. of dissolved CO2. The consequences of these changes are an increase in cathode activity, a decrease in faradaic efficiencies for H2 and CH4, and an increase in faradaic efficiencies for CO, C2H4, and EtOH, in full agreement with exptl. observations for CO2 redn. over Ag and Cu.
- 27Liu, M.; Pang, Y.; Zhang, B.; De Luna, P.; Voznyy, O.; Xu, J.; Zheng, X.; Dinh, C. T.; Fan, F.; Cao, C.; de Arquer, F. P. G.; Safaei, T. S.; Mepham, A.; Klinkova, A.; Kumacheva, E.; Filleter, T.; Sinton, D.; Kelley, S. O.; Sargent, E. H. Enhanced Electrocatalytic CO2 Reduction via Field-Induced Reagent Concentration. Nature 2016, 537 (7620), 382– 386, DOI: 10.1038/nature19060[Crossref], [PubMed], [CAS], Google Scholar27https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC28Xht1yqsbzO&md5=11d8dcf8010b105587ba93d7035bc657Enhanced electrocatalytic CO2 reduction via field-induced reagent concentrationLiu, Min; Pang, Yuanjie; Zhang, Bo; De Luna, Phil; Voznyy, Oleksandr; Xu, Jixian; Zheng, Xueli; Dinh, Cao Thang; Fan, Fengjia; Cao, Changhong; Garcia de Arquer, F. Pelayo; Safaei, Tina Saberi; Mepham, Adam; Klinkova, Anna; Kumacheva, Eugenia; Filleter, Tobin; Sinton, David; Kelley, Shana O.; Sargent, Edward H.Nature (London, United Kingdom) (2016), 537 (7620), 382-386CODEN: NATUAS; ISSN:0028-0836. (Nature Publishing Group)Electrochem. redn. of CO2 (CO2) to CO (CO) is the 1st step in the synthesis of more complex C-based fuels and feedstocks using renewable electricity. Unfortunately, the reaction suffers from slow kinetics owing to the low local concn. of CO2 surrounding typical CO2 redn. reaction catalysts. Alkali metal cations are known to overcome this limitation through noncovalent interactions with adsorbed reagent species, but the effect is restricted by the soly. of relevant salts. Large applied electrode potentials can also enhance CO2 adsorption, but this comes at the cost of increased H (H2) evolution. Here we report that nanostructured electrodes produce, at low applied overpotentials, local high elec. fields that conc. electrolyte cations, which in turn leads to a high local concn. of CO2 close to the active CO2 redn. reaction surface. Simulations reveal 10-fold higher elec. fields assocd. with metallic nm-sized tips compared to quasi-planar electrode regions, and measurements using Au nanoneedles confirm a field-induced reagent concn. that enables the CO2 redn. reaction to proceed with a geometric c.d. for CO of 22 mA per square centimeter at -0.35 V (overpotential of 0.24 V). This performance surpasses by an order of magnitude the performance of the best Au nanorods, nanoparticles and oxide-derived noble metal catalysts. Similarly designed Pd nanoneedle electrocatalysts produce formate with a faradaic efficiency of >90 per cent and an unprecedented geometric c.d. for formate of 10 mA per square centimeter at -0.2 V, demonstrating the wider applicability of the field-induced reagent concn. concept.
- 28Varela, A. S.; Kroschel, M.; Reier, T.; Strasser, P. Controlling the Selectivity of CO2 Electroreduction on Copper: the Effect of the Electrolyte Concentration and the Importance of the Local pH. Catal. Today 2016, 260, 8– 13, DOI: 10.1016/j.cattod.2015.06.009[Crossref], [CAS], Google Scholar28https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC2MXhtFeitrrE&md5=3af2d3ae954b9ce2d779503eb6bca663Controlling the selectivity of CO2 electroreduction on copper: The effect of the electrolyte concentration and the importance of the local pHVarela, Ana Sofia; Kroschel, Matthias; Reier, Tobias; Strasser, PeterCatalysis Today (2016), 260 (), 8-13CODEN: CATTEA; ISSN:0920-5861. (Elsevier B.V.)The activity and selectivity of Cu during the CO2 electrochem. redn. can be tuned by changing the concn. of the bicarbonate electrolyte. Comparing the abs. formation rate and faradaic selectivity of H2, CH4, CO, and C2H4 as a function of the applied electrode potential, variations in the bulk buffer capacities of the electrolyte have substantial impact on abs. product formation rates and relative faradaic selectivity. High concns. of bicarbonate improve the overall faradaic CO2 electroredn. activity, largely due to higher abs. formation rates of H2 and CH4. In lower-concd. bicarbonate electrolytes with their lower overall activity, the selectivity toward ethylene was drastically enhanced. Following earlier theor. work, the authors hypothesize the pH near the Cu electrode interface to largely account for the obsd. effects: dild. KHCO3 solns. allow for more alk. local pH values during CO2 electroredn. The authors' study highlights the controlling role of the interfacial pH on the product distribution during CO2 redn. over a wide electrode potential range.
- 29Verma, S.; Lu, X.; Ma, S.; Masel, R. I.; Kenis, P. J. A. The Effect of Electrolyte Composition on the Electroreduction of CO2 To CO on Ag Based Gas Diffusion Electrodes. Phys. Chem. Chem. Phys. 2016, 18 (10), 7075– 7084, DOI: 10.1039/C5CP05665A[Crossref], [PubMed], [CAS], Google Scholar29https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC2MXhvVygs7rF&md5=d489d730587522ee37bf3c28c511c77aThe effect of electrolyte composition on the electroreduction of CO2 to CO on Ag based gas diffusion electrodesVerma, Sumit; Lu, Xun; Ma, Sichao; Masel, Richard I.; Kenis, Paul J. A.Physical Chemistry Chemical Physics (2016), 18 (10), 7075-7084CODEN: PPCPFQ; ISSN:1463-9076. (Royal Society of Chemistry)The electroredn. of CO2 to C1-C2 chems. can be a potential strategy for using CO2 as a C feedstock. The authors study the effect of electrolytes on the electroredn. of CO2 to CO on Ag based gas diffusion electrodes. Electrolyte concn. was found to play a major role in the process for the electrolytes (KOH, KCl, and KHCO3) studied here. Several fold improvements in partial current densities of CO (jCO) were obsd. on moving from 0.5 M to 3.0 M electrolyte soln. independent of the nature of the anion. jCO values ≤440 mA cm-2 with an energy efficiency (EE) of ≈ 42% and 230 mA cm-2 with EE ≈ 54% were obsd. when using 3.0 M KOH. Electrochem. impedance spectroscopy showed that both the charge transfer resistance (Rct) and the cell resistance (Rcell) decreased on moving from a 0.5 M to a 3.0 M KOH electrolyte. Anions play an important role with respect to reducing the onset potential of CO in the order OH- (-0.13 V vs. RHE) < HCO3- (-0.46 V vs. RHE) < Cl- (-0.60 V vs. RHE). A decrease in Rct upon increasing electrolyte concn. and the effect of anions on the cathode can be explained by an interplay of different interactions in the elec. double layer that can either stabilize or destabilize the rate limiting CO2̇ - radical. EMIM based ionic liqs. and 1 : 2 choline Cl urea based deep eutectic solvents (DESs) were used for CO2 capture but exhibit low cond. Here, the authors study if the addn. of KCl to such solns. can improve cond. and hence jCO. Electrolytes contg. KCl in combination with EMIM Cl, choline Cl, or DESs showed a two to three fold improvement in jCO in comparison to those without KCl. Using such mixts. can be a strategy for integrating the process of CO2 capture with CO2 conversion.
- 30Higgins, D.; Hahn, C.; Xiang, C.; Jaramillo, T. F.; Weber, A. Z. Gas-Diffusion Electrodes for Carbon Dioxide Reduction: a New Paradigm. ACS Energy Lett. 2019, 4 (1), 317– 324, DOI: 10.1021/acsenergylett.8b02035[ACS Full Text], [CAS], Google Scholar30https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC1cXisFehur%252FK&md5=4eb4228156f69de1cf3f61a7011b36a9Gas-Diffusion Electrodes for Carbon Dioxide Reduction: A New ParadigmHiggins, Drew; Hahn, Christopher; Xiang, Chengxiang; Jaramillo, Thomas F.; Weber, Adam Z.ACS Energy Letters (2019), 4 (1), 317-324CODEN: AELCCP; ISSN:2380-8195. (American Chemical Society)A review. Significant advances have been made in recent years discovering new electrocatalysts and developing a fundamental understanding of electrochem. CO2 redn. processes. This field has progressed to the point that efforts can now focus on translating this knowledge toward the development of practical CO2 electrolyzers, which have the potential to replace conventional petrochem. processes as a sustainable route to produce fuels and chems. In this Perspective, we take a crit. look at the progress in incorporating electrochem. CO2 redn. catalysts into practical device architectures that operate using vapor-phase CO2 reactants, thereby overcoming intrinsic limitations of aq.-based systems. Performance comparison is made between state-of-the-art CO2 electrolyzers and com. H2O electrolyzers-a well-established technol. that provides realistic performance targets. Beyond just higher rates, vapor-fed reactors represent new paradigms for unprecedented control of local reaction conditions, and we provide a perspective on the challenges and opportunities for generating fundamental knowledge and achieving technol. progress toward the development of practical CO2 electrolyzers.
- 31Singh, M. R.; Papadantonakis, K.; Xiang, C.; Lewis, N. S. An Electrochemical Engineering Assessment of the Operational Conditions and Constraints for Solar-Driven Water-Splitting Systems at Near-Neutral pH. Energy Environ. Sci. 2015, 8 (9), 2760– 2767, DOI: 10.1039/C5EE01721A[Crossref], [CAS], Google Scholar31https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC2MXhtFSksLzN&md5=b5918f12163160e02f793d28857d186bAn electrochemical engineering assessment of the operational conditions and constraints for solar-driven water-splitting systems at near-neutral pHSingh, Meenesh R.; Papadantonakis, Kimberly; Xiang, Chengxiang; Lewis, Nathan S.Energy & Environmental Science (2015), 8 (9), 2760-2767CODEN: EESNBY; ISSN:1754-5706. (Royal Society of Chemistry)The soln. transport losses in a one-dimensional solar-driven water-splitting cell that operates in either concd. acid, dil. acid, or buffered near-neutral pH electrolytes have been evaluated using a math. model that accounts for diffusion, migration and convective transport, as well as for bulk electrochem. reactions in the electrolyte. The Ohmic resistance loss, the Nernstian potential loss assocd. with pH gradients at the surface of the electrode, and electrodialysis in different electrolytes were assessed quant. in a stagnant cell as well as in a bubble-convected cell, in which convective mixing occurred due to product-gas evolution. In a stagnant cell that did not have convective mixing, small limiting current densities (<3 mA cm-2) and significant polarization losses derived from pH gradients were present in dil. acid as well as in near-neutral pH buffered electrolytes. In contrast, bubble-convected cells exhibited a significant increase in the limiting c.d., and a significant redn. of the concn. overpotentials. In a bubble-convected cell, minimal soln. transport losses were present in membrane-free cells, in either buffered electrolytes or in unbuffered solns. with pH ≤ 1. However, membrane-free cells lack a mechanism for product-gas sepn., presenting significant practical and engineering impediments to the deployment of such systems. To produce an intrinsically safe cell, an ion-exchange membrane was incorporated into the cell. The accompanying soln. losses, esp. the pH gradients at the electrode surfaces, were modeled and simulated for such a system. Hence this work describes the general conditions under which intrinsically safe, efficient solar-driven water-splitting cells can be operated.
- 32Lobaccaro, P.; Singh, M. R.; Clark, E. L.; Kwon, Y.; Bell, A. T.; Ager, J. W., III. Effects of Temperature and Gas–Liquid Mass Transfer on the Operation of Small Electrochemical Cells for the Quantitative Evaluation of CO2 Reduction Electrocatalysts. Phys. Chem. Chem. Phys. 2016, 18 (38), 26777– 26785, DOI: 10.1039/C6CP05287H[Crossref], [PubMed], [CAS], Google Scholar32https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC28XhsVKitr3F&md5=b9b3f9bf7bbf205dddb69272df3aa009Effects of temperature and gas-liquid mass transfer on the operation of small electrochemical cells for the quantitative evaluation of CO2 reduction electrocatalystsLobaccaro, Peter; Singh, Meenesh R.; Clark, Ezra Lee; Kwon, Youngkook; Bell, Alexis T.; Ager, Joel W.Physical Chemistry Chemical Physics (2016), 18 (38), 26777-26785CODEN: PPCPFQ; ISSN:1463-9076. (Royal Society of Chemistry)In the last few years, there has been increased interest in electrochem. CO2 redn. (CO2R). Many exptl. studies employ a membrane sepd., electrochem. cell with a mini H-cell geometry to characterize CO2R catalysts in aq. soln. This type of electrochem. cell is a mini-chem. reactor and it is important to monitor the reaction conditions within the reactor to ensure that they are const. throughout the study. We show that operating cells with high catalyst surface area to electrolyte vol. ratios (S/V) at high current densities can have subtle consequences due to the complexity of the phys. phenomena taking place on electrode surfaces during CO2R, particularly as they relate to the cell temp. and bulk electrolyte CO2 concn. Both effects were evaluated quant. in high S/V cells using Cu electrodes and a bicarbonate buffer electrolyte. Electrolyte temp. is a function of the current/total voltage passed through the cell and the cell geometry. Even at a very high c.d., 20 mA cm-2, the temp. increase was less than 4 °C and a decrease of <10% in the dissolved CO2 concn. is predicted. In contrast, limits on the CO2 gas-liq. mass transfer into the cells produce much larger effects. By using the pH in the cell to measure the CO2 concn., significant undersatn. of CO2 is obsd. in the bulk electrolyte, even at more modest current densities of 10 mA cm-2. Undersatn. of CO2 produces large changes in the faradaic efficiency obsd. on Cu electrodes, with H2 prodn. becoming increasingly favored. We show that the size of the CO2 bubbles being introduced into the cell is crit. for maintaining the equil. CO2 concn. in the electrolyte, and we have designed a high S/V cell that is able to maintain the near-equil. CO2 concn. at current densities up to 15 mA cm-2.
- 33Weng, L.-C.; Bell, A. T.; Weber, A. Z. Towards Membrane-Electrode Assembly Systems for CO2 Reduction: a Modeling Study. Energy Environ. Sci. 2019, 12 (6), 1950– 1968, DOI: 10.1039/C9EE00909D[Crossref], [CAS], Google Scholar33https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC1MXptFKhu70%253D&md5=423c940774b5d165b0c50a0a0dfb6256Towards membrane-electrode assembly systems for CO2 reduction: a modeling studyWeng, Lien-Chun; Bell, Alexis T.; Weber, Adam Z.Energy & Environmental Science (2019), 12 (6), 1950-1968CODEN: EESNBY; ISSN:1754-5706. (Royal Society of Chemistry)Membrane-electrode assemblies (MEAs) are an attractive cell design for the electrochem. redn. of CO2 because they exhibit low ohmic loss and high energy efficiency. We describe here the development and application of a multiphysics model to investigate the fundamental limitations of two MEA designs: one with gaseous feeds at both the anode and cathode (full-MEA), and the other with an aq. anode feed (KHCO3 or KOH exchange soln.) and a gaseous cathode feed (exchange-MEA). The total c.d. for the three cases follows the order: KOH-MEA > KHCO3-MEA > full-MEA. This trend is established by examg. the distribution of the applied voltage. We show that the main charge-carrying species are carbonate anions for an MEA that uses an anion-exchange membrane (AEM). The amt. of CO2 consumed but not converted to CO decreases with increasing current densities above 100 mA cm-2 for a full-MEA, but converges to 50% for exchange-MEAs. The full-MEA becomes limited by ohmic resistance as the membrane dehydrates with increasing cell temp., and eventually becomes limited due to water mass transport. The exchange-MEAs can maintain membrane hydration and the local ion concn. at the anode, but are limited by salt pptn. at the cathode, as well as a higher tendency to flood. Finally, we explore the effects of temp. and discuss the possibility of increasing water supply to the full-MEA to improve its performance at elevated temps. The MEA model and the understanding of MEA performance for the electrochem. redn. of CO2 presented in this study should help guide the design of next-generation CO2 redn. cells.
- 34Song, J. T.; Song, H.; Kim, B.; Oh, J. Towards Higher Rate Electrochemical CO2 Conversion: From Liquid-Phase to Gas-Phase Systems. Catalysts 2019, 9 (3), 224, DOI: 10.3390/catal9030224[Crossref], [CAS], Google Scholar34https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC1MXotlGjtrw%253D&md5=ec441ee8bd7ad07f1d52c09e4259e31eTowards higher rate electrochemical CO2 conversion: from liquid-phase to gas-phase systemsSong, Jun Tae; Song, Hakhyeon; Kim, Beomil; Oh, JihunCatalysts (2019), 9 (3), 224/1-224/25CODEN: CATACJ; ISSN:2073-4344. (MDPI AG)Electrochem. CO2 conversion offers a promising route for value-added products such as formate, carbon monoxide, and hydrocarbons. As a result of the highly required overpotential for CO2 redn., researchers have extensively studied the development of catalyst materials in a typical H-type cell, utilizing a dissolved CO2 reactant in the liq. phase. However, the low CO2 soly. in an aq. soln. has critically limited productivity, thereby hindering its practical application. In efforts to realize com. available CO2 conversion, gas-phase reactor systems have recently attracted considerable attention. Although the achieved performance to date reflects a high feasibility, further development is still required in order for a well-established technol. Accordingly, this review aims to promote the further study of gas-phase systems for CO2 redn., by generally examg. some previous approaches from liq.-phase to gas-phase systems. Finally, we outline major challenges, with significant lessons for practical CO2 conversion systems.
- 35Li, J.; Chen, G.; Zhu, Y.; Liang, Z.; Pei, A.; Wu, C.-L.; Wang, H.; Lee, H. R.; Liu, K.; Chu, S.; Cui, Y. Efficient Electrocatalytic CO2 Reduction on a Three-Phase Interface. Nature Catalysis 2018, 1 (8), 592– 600, DOI: 10.1038/s41929-018-0108-3[Crossref], [CAS], Google Scholar35https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC1MXhtFGisb7I&md5=d2fb2bf7cb72e2e88d22a4fdbd8d2700Efficient electrocatalytic CO2 reduction on a three-phase interfaceLi, Jun; Chen, Guangxu; Zhu, Yangying; Liang, Zheng; Pei, Allen; Wu, Chun-Lan; Wang, Hongxia; Lee, Hye Ryoung; Liu, Kai; Chu, Steven; Cui, YiNature Catalysis (2018), 1 (8), 592-600CODEN: NCAACP; ISSN:2520-1158. (Nature Research)Electrochem. CO2 redn. is a crit. approach to reducing the globally accelerating CO2 emission and generating value-added products. Despite great efforts to optimize catalyst activity and selectivity, facilitating the catalyst accessibility to high CO2 concns. while maintaining electrode durability remains a significant challenge. Here, we designed a catalytic system that mimics the alveolus structure in mammalian lungs with high gas permeability but very low water diffusibility, enabling an array of three-phase catalytic interfaces. Flexible, hydrophobic, nanoporous polyethylene membranes with high gas permeability were used to enable efficient CO2 access and a high local alky. on the catalyst surface at different CO2 flow rates. Such an alveolus-mimicking structure generates a high CO prodn. Faradaic efficiency of 92% and excellent geometric current densities of CO prodn. (25.5 mA cm-2) at -0.6 V vs. the reversible hydrogen electrode, with a very thin catalyst thickness of 20-80 nm.
- 36Weng, L.-C.; Bell, A. T.; Weber, A. Z. Modeling Gas-Diffusion Electrodes for CO2 Reduction. Phys. Chem. Chem. Phys. 2018, 20, 16973– 16984, DOI: 10.1039/C8CP01319E[Crossref], [PubMed], [CAS], Google Scholar36https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC1cXhtVKntb%252FI&md5=d5beb1df920eac710ede8615667d7e11Modeling gas-diffusion electrodes for CO2 reductionWeng, Lien-Chun; Bell, Alexis T.; Weber, Adam Z.Physical Chemistry Chemical Physics (2018), 20 (25), 16973-16984CODEN: PPCPFQ; ISSN:1463-9076. (Royal Society of Chemistry)CO2 redn. conducted in electrochem. cells with planar electrodes immersed in an aq. electrolyte is severely limited by mass transport across the hydrodynamic boundary layer. This limitation can be minimized by use of vapor-fed, gas-diffusion electrodes (GDEs), enabling current densities that are almost two orders of magnitude greater at the same applied cathode overpotential than what is achievable with planar electrodes in an aq. electrolyte. The addn. of porous cathode layers, however, introduces a no. of parameters that need to be tuned in order to optimize the performance of the GDE cell. In this work, we develop a multiphysics model for gas diffusion electrodes for CO2 redn. and used it to investigate the interplay between species transport and electrochem. reaction kinetics. The model demonstrates how the local environment near the catalyst layer, which is a function of the operating conditions, affects cell performance. We also examine the effects of catalyst layer hydrophobicity, loading, porosity, and electrolyte flowrate to help guide exptl. design of vapor-fed CO2 redn. cells.
- 37Cook, R. L.; MacDuff, R. C.; Sammells, A. F. High Rate Gas Phase CO2 Reduction to Ethylene and Methane Using Gas Diffusion Electrodes. J. Electrochem. Soc. 1990, 137 (2), 607– 608, DOI: 10.1149/1.2086515[Crossref], [CAS], Google Scholar37https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADyaK3cXhsVOkurw%253D&md5=0fbbb2a641aaed5ebaf1cd904242cf33High rate gas phase carbon dioxide reduction to ethylene and methane using gas diffusion electrodesCook, Ronald L.; MacDuff, Robert C.; Sammells, Anthony F.Journal of the Electrochemical Society (1990), 137 (2), 607-8CODEN: JESOAN; ISSN:0013-4651.Gas phase redn. of CO2 was studied at gas diffusion electrodes made from Cu gauge with C black and Teflon with and without supported Cu. Cu is the active site for CO2 redn. Ethylene was the dominant redn. product. Faradaic efficiencies of 71.3% were obtained for gas phase redn. of CO2 to hydrocarbon at c.d. >0.5 A/cm2 using the gas diffusion electrode.
- 38Hori, Y.; Ito, H.; Okano, K.; Nagasu, K.; Sato, S. Silver-Coated Ion Exchange Membrane Electrode Applied to Electrochemical Reduction of Carbon Dioxide. Electrochim. Acta 2003, 48 (18), 2651– 2657, DOI: 10.1016/S0013-4686(03)00311-6[Crossref], [CAS], Google Scholar38https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BD3sXltFCmsb4%253D&md5=65df35bb823ad0fbf9820b1cbd560e68Silver-coated ion exchange membrane electrode applied to electrochemical reduction of carbon dioxideHori, Y.; Ito, H.; Okano, K.; Nagasu, K.; Sato, S.Electrochimica Acta (2003), 48 (18), 2651-2657CODEN: ELCAAV; ISSN:0013-4686. (Elsevier Science B.V.)Silver-coated ion exchange membrane electrodes (solid polymer electrolyte, SPE) were prepd. by electroless deposition of silver onto ion exchange membranes. The SPE electrodes were used for carbon dioxide (CO2) redn. with 0.2 M K2SO4 as the electrolyte with a platinum plate (Pt) for the counterelectrode. In an SPE electrode system prepd. from a cation exchange membrane (CEM), the surface of the SPE was partly ruptured during CO2 redn., and the reaction was rapidly suppressed. SPE electrodes made of an anion exchange membrane (SPE/AEM) sustained redn. of CO2 to CO for more than 2 h, whereas, the electrode potential shifted neg. during the electrolysis. The reaction is controlled by the diffusion of CO2 through the metal layer of the SPE electrode at high c.d. Ultrasonic radiation, applied to the prepn. of SPE/AEM, was effective to improve the electrode properties, enhancing the electrolysis current of CO2 redn. Observation by a scanning electron microscope (SEM) showed that the electrode metal layer became more porous by the ultrasonic radiation treatment. The partial c.d. of CO2 redn. by SPE/AEM amounted to 60 mA cm-2, i.e. three times the upper limit of the conventional electrolysis by a plate electrode. Application of SPE device may contribute to an advancement of CO2 fixation at ambient temp. and pressure.
- 39CRC Handbook of Chemistry and Physics; Lide, D. R., Ed.; New York, 2003.
- 40Ripatti, D. S.; Veltman, T. R.; Kanan, M. W. Carbon Monoxide Gas Diffusion Electrolysis That Produces Concentrated C2 Products with High Single-Pass Conversion. Joule 2019, 3 (1), 240– 256, DOI: 10.1016/j.joule.2018.10.007[Crossref], [CAS], Google Scholar40https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC1MXhsValt7c%253D&md5=d301c4721ed676d3fe66d8524ee6e8d1Carbon Monoxide Gas Diffusion Electrolysis that Produces Concentrated C2 Products with High Single-Pass ConversionRipatti, Donald S.; Veltman, Thomas R.; Kanan, Matthew W.Joule (2019), 3 (1), 240-256CODEN: JOULBR; ISSN:2542-4351. (Cell Press)Electrochem. CO conversion is crit. for the development of alternative fuel and chem. syntheses. To be efficient, electrosynthesis must make concd. product streams at high rates with modest potentials, but the combination of these features has not been established for CO or the related CO2 electrolysis. Here we investigate CO electrolysis with gas diffusion electrodes (GDEs) supplied by interdigitated flow fields in electrochem. cells with different ion transport properties. By optimizing gas and ion transport, we show that it is possible to simultaneously achieve high c.d., high selectivity, and high single-pass conversion at moderate cell potentials. Using a cell with the GDE directly contacting a Nafion membrane, we demonstrate >100 mA cm-2 CO redn. to C2 products and direct prodn. of 1.1 M acetate at a cell potential of 2.4 V over 24 h. Our results reveal crit. design features for maximizing the efficiency of C2 electrosynthesis.
- 41Dinh, C. T.; Burdyny, T.; Kibria, M. G.; Seifitokaldani, A.; Gabardo, C. M.; de Arquer, F. P. G.; Kiani, A.; Edwards, J. P.; De Luna, P.; Bushuyev, O. S.; Zou, C.; Quintero-Bermudez, R.; Pang, Y.; Sinton, D.; Sargent, E. H. CO2 Electroreduction to Ethylene via Hydroxide-Mediated Copper Catalysis at an Abrupt Interface. Science 2018, 360 (6390), 783– 787, DOI: 10.1126/science.aas9100[Crossref], [PubMed], [CAS], Google Scholar41https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC1cXpsVCgsL0%253D&md5=0beec1cdcc8939b3eb057cb6b26742f6Carbon dioxide electroreduction to ethylene via hydroxide-mediated copper catalysis at an abrupt interfaceDinh, Cao-Thang; Burdyny, Thomas; Kibria, Md Golam; Seifitokaldani, Ali; Gabardo, Christine M.; Garcia de Arquer, F. Pelayo; Kiani, Amirreza; Edwards, Jonathan P.; De Luna, Phil; Bushuyev, Oleksandr S.; Zou, Chengqin; Quintero-Bermudez, Rafael; Pang, Yuanjie; Sinton, David; Sargent, Edward H.Science (Washington, DC, United States) (2018), 360 (6390), 783-787CODEN: SCIEAS; ISSN:0036-8075. (American Association for the Advancement of Science)Carbon dioxide (CO2) electroredn. could provide a useful source of ethylene, but low conversion efficiency, low prodn. rates, and low catalyst stability limit current systems. Here we report that a copper electrocatalyst at an abrupt reaction interface in an alk. electrolyte reduces CO2 to ethylene with 70% faradaic efficiency at a potential of -0.55 V vs. a reversible hydrogen electrode (RHE). Hydroxide ions on or near the copper surface lower the CO2 redn. and carbon monoxide (CO)-CO coupling activation energy barriers; as a result, onset of ethylene evolution at -0.165 V vs. an RHE in 10 M potassium hydroxide occurs almost simultaneously with CO prodn. Operational stability was enhanced via the introduction of a polymer-based gas diffusion layer that sandwiches the reaction interface between sep. hydrophobic and conductive supports, providing const. ethylene selectivity for an initial 150 operating hours.
- 42Li, Y. C.; Lee, G.; Yuan, T.; Wang, Y.; Nam, D.-H.; Wang, Z.; Garcia de Arquer, F. P.; Lum, Y.; Dinh, C. T.; Voznyy, O.; Sargent, E. H. Co2 Electroreduction From Carbonate Electrolyte. ACS Energy Lett. 2019, 4 (6), 1427– 1431, DOI: 10.1021/acsenergylett.9b00975[ACS Full Text], [CAS], Google Scholar42https://chemport.cas.org/services/resolver?origin=ACS&resolution=options&coi=1%3ACAS%3A528%3ADC%252BC1MXhtVejtrfF&md5=54aa3a6b76f98e7735a2748b2eb9de57CO2 Electroreduction from Carbonate ElectrolyteLi, Yuguang C.; Lee, Geonhui; Yuan, Tiange; Wang, Ying; Nam, Dae-Hyun; Wang, Ziyun; Garcia de Arquer, F. Pelayo; Lum, Yanwei; Dinh, Cao-Thang; Voznyy, Oleksandr; Sargent, Edward H.ACS Energy Letters (2019), 4 (6), 1427-1431CODEN: AELCCP; ISSN:2380-8195. (American Chemical Society)The process of CO2 valorization-from capture of CO2 to its electrochem. upgrade-requires significant inputs in each of the capture, upgrade, and sepn. steps. Here the authors report an electrolyzer that upgrades carbonate electrolyte from CO2 capture soln. to syngas, achieving 100% C use across the system. A bipolar membrane was used to produce proton in situ to facilitate CO2 release at the membrane:catalyst interface from the carbonate soln. Using a Ag catalyst, the authors generate syngas at a 3:1 H2:CO ratio, and the product is not dild. by CO2 at the gas outlet; the authors generate this pure syngas product stream at a c.d. of 150 mA/cm2 and an energy efficiency of 35%. The carbonate-to-syngas system is stable under a continuous 145 h of catalytic operation. The work demonstrates the benefits of coupling CO2 electrolysis with a CO2 capture electrolyte on the path to practicable CO2 conversion technologies.
- 43Hossain, M. Z.; Rahim, N. A.; Selvaraj, J. A. L. Recent Progress and Development on Power DC-DC Converter Topology, Control, Design and Applications: a Review. Renewable Sustainable Energy Rev. 2018, 81, 205– 230, DOI: 10.1016/j.rser.2017.07.017
Supporting Information
ARTICLE SECTIONSThe Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsenergylett.9b02576.
Methods, calculations of solar-to-fuel efficiency, GDE efficiency, turnover frequency, cell potentials, CO2 loss, and supporting figures and tables (PDF)
Terms & Conditions
Electronic Supporting Information files are available without a subscription to ACS Web Editions. The American Chemical Society holds a copyright ownership interest in any copyrightable Supporting Information. Files available from the ACS website may be downloaded for personal use only. Users are not otherwise permitted to reproduce, republish, redistribute, or sell any Supporting Information from the ACS website, either in whole or in part, in either machine-readable form or any other form without permission from the American Chemical Society. For permission to reproduce, republish and redistribute this material, requesters must process their own requests via the RightsLink permission system. Information about how to use the RightsLink permission system can be found at http://pubs.acs.org/page/copyright/permissions.html.