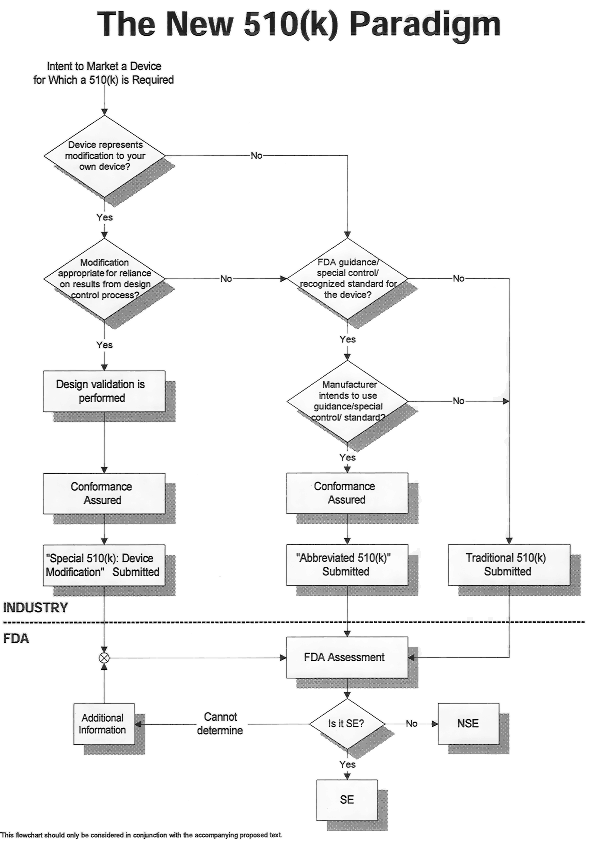
An applicant may choose from three types of Premarket Notification 510(k) submissions for marketing clearance: Traditional, Special, and Abbreviated.
Traditional method is the original complete submission as provided in 21 CFR 807. In 1998, FDA developed "The New 510(k) Paradigm" to streamline the evaluation of Premarket Notifications. The New 510(k) Paradigm provides two optional approaches to the Traditional 510(k) method for obtaining 510(k) marketing clearance under certain instances: Special 510(k) and Abbreviated 510(k). The Special 510(k): Device Modification utilizes certain aspects of the Quality System Regulation and the Abbreviated 510(k) relies on the use of guidance documents, special controls, and recognized standards to facilitate 510(k) review.
The use of either alternative does not affect FDA's ability to obtain any information authorized by the statute or regulations. Additional guidance on the new 510(k) paradigm can be found in Frequently Asked Questions on the New 510(k) Paradigm.
The following reference flowchart below outlines which type of 510(k) is suitable for a given set of circumstances.
The 510(k) Review Fee is the same for Traditional, Special, and Abbreviated 510(k)s.
Form FDA-3674, ClinicalTrials.gov Data Bank
Title VIII of the Food and Drug Administration Amendments Act of 2007 (FDAAA) included a provision that all 510(k) submissions are required to be accompanied with certification that all applicable clinical trial information has been submitted to the ClinicalTrials.gov data bank.
If your 510(k) includes data from a clinical trial, you must determine if your study is applicable for entry into the clinical trial registry data bank at ClinicalTrials.gov. Based on this determination, check box 9.B. or 9.C., and complete the applicable sections of the form. An applicable device clinical trial is a prospective clinical study of health outcomes
comparing an intervention with a device
against a
control in human subjects (other than a small clinical
trial to determine the feasibility of a device,
or a clinical trial to test prototype devices where
the primary outcome measure relates to feasibility
and not to health outcomes). See Title VIII - Clinical Trial Databases. Currently, FDA is reviewing the legislation and developing guidance on which clinical trials meet the definition of "applicable" trials and are required to report to ClinicalTrials.gov. Until FDA issues this guidance, 510(k) submitters are responsible for determining whether their studies meet the definition of an applicable trial and, therefore, are subject to reporting requirements.
Information on how to register your clinical trial(s) in the ClinicalTrials.gov data bank is available on the National Library of Medicine (NLM) Protocol Registration System (PRS) website.
Traditional 510(k)
The Traditional 510(k) may used for any original 510(k) or for a modification to a previously cleared device under 510(k). The traditional method is the original complete submission as provided in 21 CFR 807. The Traditional 510(k) method may be used under any circumstances.
The Special 510(k) is used for device modifications and utilizes the design controls aspect of the Quality System (QS) regulation (21 CFR 820.30). Special 510(k)s may be submitted for a modification to a device that has been cleared under the 510(k) process. If a new 510(k) is needed for the modification and if the modification does not affect the intended use of the device or alter the fundamental scientific technology of the device, then summary information that results from the design control process can serve as the basis for clearing the application.
Under the Quality System regulation, all Class II and III devices and certain Class I devices are required to be designed in conformance to section 820.30 Design Controls. The Special 510(k) allows the manufacturer to declare conformance to design controls without providing the data. Manufacturers of Class I devices requiring 510(k) may elect to comply with the design control provision of the QS regulation and submit a Special 510(k).
Under the Special 510(k) option, 510(k) holders who intend to modify their own legally marketed device will conduct the risk analysis and the necessary verification and validation activities to demonstrate that the design outputs of the modified device meet the design input requirements. Once the 510(k) holder has ensured the satisfactory completion of this process, a "Special 510(k): Device Modification" may be submitted. While the basic content requirements of the 510(k) (21 CFR 807.87) will remain the same, this type of submission should also reference the cleared 510(k) number and contain a "Declaration of Conformity" with design control requirements.
In order to provide an incentive for 510(k) holders to choose this option for obtaining FDA clearance for device modifications, the Office of Device Evaluation (ODE) and Office of In Vitro Diagnostic Device Evaluation and Safety (OIVD) intend to process Special 510(k)s within 30 days of receipt by FDAs Document Mail Center (DMC).
Some device modifications may be implemented without submission of a new 510(k). The submitter should review Is a new 510(k) required for a modification to the device? to assure that a new 510(k) is required for the modification to the device.
Device manufacturers may choose to submit an Abbreviated 510(k) when:
a guidance documents exists,
a special control has been established, or
FDA has recognized a relevant consensus standard.
An Abbreviated 510(k) submission must include the required elements identified in 21 CFR 807.87 [Traditional 510(k)]. However, in an Abbreviated 510(k) submission, firms elect to provide summary reports on the use of guidance documents and/or special controls or declarations of conformity to FDA recognized standards to expedite the review of a submission.