FDA
Home Page | CDRH Home Page | Search
| CDRH
A-Z Index | Contact CDRH
![]()
|
| FDA
Home Page | CDRH Home Page | Search
| CDRH
A-Z Index | Contact CDRH
|
|
|
|||||||||||||||||||||
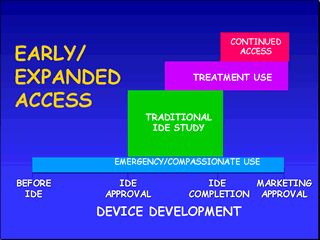
Early/Expanded AccessAn unapproved medical device may normally only be used on human subjects through an approved clinical study in which the subjects meet certain criteria and the device is only used in accordance with the approved protocol by a clinical investigator participating in the clinical trial. However, there may be circumstances under which a health care provider may wish to use an unapproved device to save the life of a patient or to help a patient suffering from a serious disease or condition for which there no other alternative therapy exists. Patients/physicians faced with these circumstances may have access to investigational devices under one of four main mechanisms by which FDA may make an unapproved device available:
These mechanisms can be utilized during a certain time-frame in the IDE process if the criteria are met. FDA approval is required except in the case of emergency use. The mechanisms are summarized below followed by an in depth discussion of criteria and requirements.
Emergency situations may arise in which there will be a need to use an investigational device in a manner inconsistent with the approved investigational plan or by a physician who is not part of the clinical study. Emergency use of an unapproved device may occur before an IDE is approved. Criteria:
Time-frame: Before or after initiation of the clinical trial There are special cases under emergency research in which the human subject is in a life-threatening situation and it is not feasible to obtain informed consent. In order to allow such research to proceed, special provisions for exception from informed consent requirements must be met. In addition, the IRB and a physician not participating in the investigation must review and approve the investigation. The sponsor must also submit a separate IDE application to FDA. Detailed information on conditions for exception from informed consent can be found under Informed Consents, Exception from Informed Consent Requirements for Emergency Research. Compassionate Use (or Single Patient/Small Group Access) The compassionate use provision allows access for patients who do not meet the requirements for inclusion in the clinical investigation but for whom the treating physician believes the device may provide a benefit in treating and/or diagnosing their disease or condition. This provision is typically approved for individual patients but may be approved to treat a small group. Criteria:
Time-frame: During clinical trial FDA recognizes that there are circumstances in which an investigational device is the only option available for a patient faced with a serious, albeit not life-threatening, disease or condition. In these circumstances, FDA uses its regulatory discretion in determining whether such use of an investigational device should occur. Prior FDA approval is needed before compassionate use occurs. In order to obtain Agency approval, the sponsor should submit an IDE supplement requesting approval for a protocol deviation under section §812.35(a) in order to treat the patient. The IDE supplement should include:
The physician should not treat the patient identified in the supplement until FDA approves use of the device under the proposed circumstances. In reviewing this type of request, FDA will consider the above information as well as whether the preliminary evidence of safety and effectiveness justifies such use and whether such use would interfere with the conduct of a clinical trial to support marketing approval. If the request is approved, the attending physician should devise an appropriate schedule for monitoring the patient, taking into consideration the investigational nature of the device and the specific needs of the patient. The patient should be monitored to detect any possible problems arising from the use of the device. Following the compassionate use of the device, a follow-up report should be submitted to FDA as an IDE supplement in which summary information regarding patient outcome is presented. If any problems occurred as a result of device use, these should be discussed in the supplement and reported to the reviewing IRB as soon as possible. The above compassionate use criteria and procedures can also be applied
when a physician wishes to treat a few patients rather than an individual patient
suffering from a serious disease or condition for which no alternative therapy adequately
meets their medical need. In this case, the physician should request access to the
investigational device through the IDE sponsor. The sponsor should submit an IDE
supplement that includes the information identified above and indicates the number of
patients to be treated. Such a supplement should include the protocol to be followed or
identify deviations from the approved clinical protocol. As with single patient
compassionate use, a monitoring schedule should be designed to meet the needs of the
patients while recognizing the investigational nature of the device. Follow-up information
on the use of the device should be submitted in an IDE supplement after all compassionate
use patients have been treated. An approved IDE specifies the maximum number of clinical sites and the maximum number of human subjects that may be enrolled in the study. During the course of the clinical trial, if the data suggests that the device is effective, then the trial may be expanded to include additional patients with life-threatening or serious diseases. Criteria:
Time-frame: During clinical trial A device that is not approved for marketing may be under clinical investigation for a serious or immediately life- threatening disease or condition in patients for whom no comparable or satisfactory alternative device or other therapy is available. During the clinical trial or prior to final action on the marketing application, it may be appropriate to use the device in the treatment of patients not in the trial under the provisions of the treatment investigational device exemptions (IDE) regulation. (§812.36) The treatment use provision of the IDE facilitates the availability of promising new devices to desperately ill patients as early in the device development process as possible, before general marketing begins, and to obtain additional data on the device's safety and effectiveness. In the case of a serious disease, a device ordinarily may be made available for treatment use under this section after all clinical trials have been completed. In the case of an immediately life-threatening disease, a device may be made available for treatment use under this section prior to the completion of all clinical trials. An ``immediately life-threatening'' disease means a stage of a disease in which there is a reasonable likelihood that death will occur within a matter of months or in which premature death is likely without early treatment. ``Treatment use'' of a device includes the use of a device for diagnostic purposes. FDA would consider the use of an investigational device under a treatment IDE if:
Applications for treatment use A treatment IDE application must include, in the following order:
A licensed practitioner who receives an investigational device for treatment use under a treatment IDE is an "investigator" under the IDE and is responsible for meeting all applicable investigator responsibilities under 21 CFR 812, 21 CFR 50, and 21 CFR 56. Applications should be identified on the outside envelope as a treatment IDE application and reference the IDE number. The original and two copies should be mailed to the following address: Food and Drug Administration FDA action on treatment IDE applications Approval of treatment IDE's Treatment use may begin 30 days after FDA receives the treatment IDE submission. FDA may notify the sponsor in writing earlier than the 30 days that the treatment use may or may not begin. FDA may approve the treatment use as proposed or approve it with modifications. Disapproval or withdrawal of approval of treatment IDE's FDA may disapprove or withdraw approval of a treatment IDE if:
Notice of disapproval or withdrawal If FDA disapproves or proposes to withdraw approval of a treatment IDE, FDA will follow the procedures set forth in the IDE regulations [§812.30(c)] For more information, see Approval Process, FDA Actions. Safeguards Treatment use of an investigational device is conditioned upon the sponsor and investigators complying with the safeguards of the IDE process and the regulations governing informed consent (21 CFR 50) and institutional review boards (21 CFR 56). Reporting requirements The sponsor of a treatment IDE must submit progress reports on a semi-annual basis to all reviewing IRB's and FDA until the filing of a marketing application. The date of these reports are based on the period of time since initial approval of the treatment IDE. After filing of a marketing application, progress reports must be submitted annually in accordance with the IDE regulation. See "Suggested Format For IDE Progress Report" under Reports for guidance on the content of a progress report. The progress report must also include the number of patients treated with the device under the treatment IDE, the names of the investigators participating in the treatment IDE, and a brief description of the sponsor's efforts to pursue marketing approval/ clearance of the device. The sponsor of a treatment IDE is responsible for submitting all other reports required under §812.150 (Reports), such as unanticipated adverse device effects and final reports. The reports are submitted as supplements to the original IDE application. See Reports for additional guidance. FDA may allow continued enrollment of subjects after the controlled clinical trial under an IDE has been completed in order to allow access to the investigational medical device while the marketing application is being prepared by the sponsor or reviewed by FDA. Criteria:
Time-frame: After completion of the clinical trial The sponsor of a clinical investigation is permitted to continue to enroll subjects while a marketing application is being prepared by the sponsor and/or reviewed by the Agency if there is:
The continued enrollment of subjects in an investigation while a marketing application is being prepared by the sponsor and/or reviewed by ODE is known as an "extended investigation." Extended investigations permit patients and/or physicians continued access to the devices while also allowing the collection of additional safety and effectiveness data to support the marketing application or to address new questions regarding the investigational device. The Continued Access Policy may be applied to any clinical investigation that meets the criteria identified above; however, it is intended to be applied late in the device development process, i.e., after the controlled clinical trial has been completed. A sponsor's request for an extended investigation should be submitted as an IDE supplement and include the following information:
There is significant overlap between the treatment IDE regulation and the Continued Access Policy. Both the Continued Access Policy and the treatment IDE regulation are intended to provide additional access to an unapproved device, once preliminary evidence regarding safety and effectiveness is available to FDA. However, because a treatment IDE can be submitted earlier in the IDE process, i.e., once promising evidence of safety and effectiveness has been collected under the IDE but while the clinical study is ongoing, it could provide access to a wider group of patients at an earlier stage in the IDE process. The treatment IDE regulation also has a more narrow application than the Continued Access Policy in that treatment use is intended to address only those patients who have an immediately life-threatening or serious disease or condition whereas the Continued Access Policy, which is applied after completion of the clinical trial, may be considered for any clinical investigation. References: Guidance on IDE Policies and
Procedures "Continued
Access to Investigational Devices During PMA Preparation and Review," Blue
Book Memorandum, D96-1 IRB
Information Sheets: Emergency Use of Unapproved Medical Devices Last modified date: July 21, 2003 |
|||||||||||||||||||||
![]()
CDRH Home Page | CDRH A-Z Index | Contact CDRH | Accessibility | Disclaimer
FDA Home Page | Search FDA Site | FDA A-Z Index | Contact FDA | HHS Home Page
Center for Devices and Radiological Health / CDRH