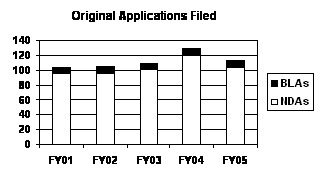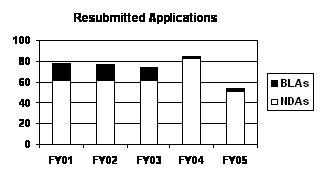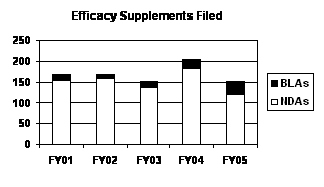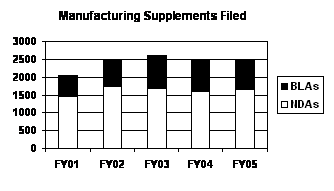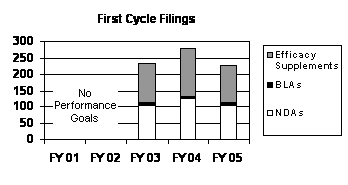|
 |
 |
|
 |
FDA Home Page | Search
FDA Site | FDA A-Z Index | Contact
FDA | FDA Centennial
![]()

 |
 |
Printable version of this report [455 KB PDF]
This page contains links to documents in Portable Document
Format (PDF). To view or
print these documents, you must use the Adobe Acrobat viewer.
Acrobat
is free and available directly from Adobe's website with full installation
instructions.
I am pleased to present the Food and Drug Administration's (FDA's) fiscal year (FY) 2005 Performance Report to the President and the Congress for the Prescription Drug User Fee Act (PDUFA). This report marks the thirteenth year of PDUFA, and completion of the third of the 5 years of the most recent reauthorization. Resources provided to FDA under PDUFA have been instrumental in new drugs reaching consumers in a more timely manner.
PDUFA I (FY 1993 through FY 1997) challenged FDA with goals to speed FDA review of new drug applications (NDAs) and biologics licensing applications (BLAs) without compromising safety. Over the course of PDUFA I, FDA exceeded all of its review performance goals. PDUFA II (FY 1998 through FY 2002) added goals to improve the process of new drug development before submission of the NDA or BLA. Under PDUFA II, most review times were shortened and FDA met or exceeded nearly all its review performance goals.
PDUFA III (FY 2003 through FY 2007) expanded fee funding to support FDA postmarket risk management and established several initiatives to improve application submissions and FDA-sponsor interactions during drug development and application review. Early and more frequent consultation with FDA helps sponsors improve the quality of their drug development and related NDAs. Under PDUFA III, FDA continues to meet most of the review performance goals. However, FDA has not been able to meet the performance targets for meetings or special protocol assessments. FDA has experienced a dramatic increase in company requests for meetings and special protocol assessments that began when the PDUFA procedural and processing goals were instituted during PDUFA II. While these FDA-sponsor interactions are important to improving drug quality, they are also imposing significant
additional work for FDA, particularly in the Center for Drug Evaluation and Research (CDER). The current user fee formulas account for adjustments in annual workload increases; however, these calculations do not take into account the disproportionate incr eases in either of these activities.
With PDUFA III expiring in September 2007, the reauthorization of PDUFA is essential to maintain the resources required to sustain the advances made in FDA review performance and to continue to advance biomedical progress.
Andrew C. von Eschenbach, M.D.
Acting Commissioner of Food and Drugs
This report presents the Food and Drug Administration's (FDA's) performance in meeting the Prescription Drug User Fee Act (PDUFA) review goals. Review performance for applications and submissions received in FY 2004 is updated and finalized. FDA's progress in meeting the quantifiable PDUFA review performance goals for FY 2004 and FY 2005 submissions and the FY 2005 procedural and processing goals are covered in this report. Additionally, this report describes FDA's progress in accomplishing new management initiatives and in meeting the information technology commitments of PDUFA III.
With all but two of the original applications submitted during FY 2004 having been reviewed and acted on by September 30, 2005 , FDA can report that it exceeded all the review performance goals for FY 2004. This occurred during a year when workload for most submissions had increased.
In FY 2005, the number of priority original applications increased for the fifth consecutive year. Priority applications represent significant new treatments. However, most other application submission categories decreased from FY 2004 to FY 2005. Submission categories that decreased included standard original applications and original efficacy supplements. The number of manufacturing supplements submitted in FY 2005 was approximately the same as in FY 2004. It is too soon to present final FY 2005 review performance; however, preliminary performance for actions completed through the end of FY 2005 is provided in each goal category.
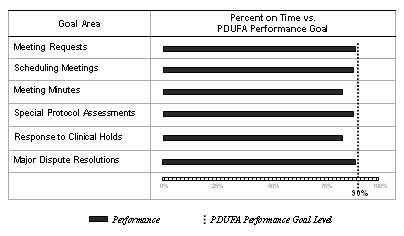
Workload related to most of the procedural and processing goals increased again in FY 2005, continuing the dramatic growth trend under PDUFA III. Meeting requests, for example, increased by 46 percent (1,662 to 2,430) from FY 2001 to FY 2005. Yearly increases in meeting requests resulted in additional increases in meeting scheduling and meeting minutes. As a result, FDA workload in all three activities increased each year between FY 2001 and FY 2005. Special protocol assessment requests also increased in FY 2005 and have increased by well over 200 percent (125 to 392) during the same
5-year period. These increases are significant because FDA must use the same staff to provide responses that are used to review applications and submissions. FDA performance related to most of the procedural and processing goals was just below the 90 percent performance levels for FY 2005.
FDA also continued to make progress on the PDUFA III Management Initiatives and Electronic Applications and Submissions commitments designed to improve the overall review process.
![]()
In 1992, Congress passed PDUFA, authorizing FDA to collect fees from companies that produce and submit applications for marketing human drug and biological products. The original PDUFA had a 5-year life; it ended in 1997, the same year Congress passed the FDA Modernization Act (FDAMA). FDAMA contained a 5-year reauthorization of PDUFA (PDUFA II) that ended on September 30, 2002 . When Congress passed the Public Health Security and Bioterrorism Preparedness and Response Act of 2002 (the Bioterrorism Act), it extended the PDUFA program for 5 more years (PDUFA III). Information about PDUFA III, including the text of the amendments and the performance goals and procedures, can be found at http://www.fda.gov/oc/pdufa/PDUFA3.html.
PDUFA requires FDA to submit two annual reports to the President and the Congress for each fiscal year during which fees are collected: 1) a performance report due within 60 days of the end of the fiscal year, and 2) a financial report due within 120 days of the end of the fiscal year. This document addresses the first of these requirements for FY 2005. This year's report covers FDA's progress in meeting the quantifiable PDUFA review goals for FY 2004 and FY 2005 submissions and the FY 2005 procedural and processing goals. The report also describes FDA's progress in accomplishing new management initiatives and in meeting the information technology commitments of PDUFA III.
PDUFA provides FDA revenue to hire additional reviewers and support staff and upgrade its information technology systems to speed up the application review process for new drugs and biological products without compromising FDA's traditionally high standards for approval. Under PDUFA, FDA is committed to achieve certain performance goals that apply to the review of original and resubmitted new product applications and efficacy and manufacturing supplements to approved applications. FDA is also committed to achieve certain procedural and processing goals aimed at facilitating and assuring quality in new drug development.
During the first few years of PDUFA I, FDA eliminated backlogs of original applications and supplements that had formed in earlier years when the program had fewer resources. Over the course of PDUFA I, FDA agreed to review and act on a progressively increasing proportion of original NDAs, BLAs, and efficacy supplements within 12 months and resubmissions and manufacturing supplements within 6 months. The FDA also agreed to review and act on 90 percent of priority NDAs, BLAs, and efficacy supplements (submissions that are for products providing significant therapeutic gains) submitted in FY 1997 within 6 months. Over the course of PDUFA I, FDA exceeded all of these performance goals.
In 1997, Congress passed FDAMA and reauthorized PDUFA (PDUFA II) for 5 more years. Under PDUFA II, most review times were shortened and FDA met or exceeded nearly all its review goals. PDUFA II expanded the scope of PDUFA work by including new goals intended to improve communication between FDA and application sponsors during the drug development process. These goals specified time frames for scheduling meetings, responding to various sponsor submissions, such as special protocols and responses to clinical holds, and other activities.
In 2002, Congress passed the Bioterrorism Act, which included an extension of PDUFA (PDUFA III) for 5 more years, FY 2003 through FY 2007. PDUFA III review performance goals and the procedural and processing goals are largely the same as the PDUFA II FY 2002 performance levels for these goals. PDUFA III establishes several new initiatives to improve application submissions and FDA-sponsor interactions during drug development and application review. In addition, it authorizes FDA to spend user fee funds on certain aspects of postmarket risk management, including surveillance of products approved after October 1, 2002 , for up to 3 years.
PDUFA-enabled improvements in review efficiency and application quality have had an impact on the overall time to marketing approval. FDA tracks a variety of metrics related to the process of human drug review. The time-to-approval statistics are affected by a number of factors, including the total number of NDA and BLA submissions as well as the overall quality of submitted applications, the number of newly submitted priority applications, and the number of review staff relative to the review workload. These factors can vary from year to year; the charts that follow provide an update on trends in submissions and overall approval times.
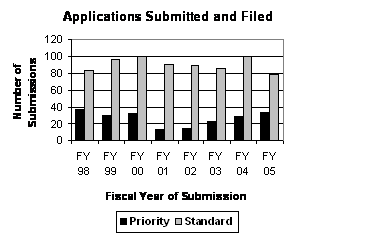
Number of FY 2005 Priority Applications Highest Since FY 1998. Priority applications represent significant therapeutic gains, and in FY 2005 they accounted for over 30 percent of the total number of original receipts. The number of priority applications increased from 29 in FY 2004 to 34 in FY 2005, continuing a 4-year trend (see graph to the right) . Standard applications submitted in FY 2005 were down from the peak year of FY 2004, to the lowest level since FY 1998
. D
D
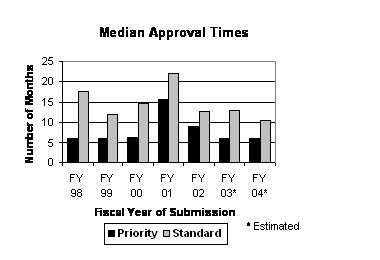
Median Time to Approval Remained Steady in FY 2004 for Priority Applications and Decreased for Standard Applications. Median approval times for priority applications decreased in FY 2002 and FY 2003 and preliminary estimates indicate that FY 2004 will maintain this level (see graph to the right). Based on applications approved through September 30, 2005 , and historical data indicating close to 80 percent of all filed applications will eventually be approved, the estimated median approval time for priority applications for FY 2003 and FY 2004 is approximately 6.0 months. The median approval time for standard applications is estimated to be 10.5 months in FY 2004, down from an estimated 13.0 months for FY 2003.
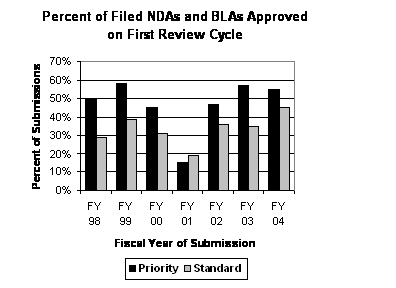
Percentage of First Cycle Approvals for Standard Applications Increased in FY 2004. The percentage of standard applications that were approved in the first cycle increased from 35 percent in FY 2003 to 45 percent in FY 2004 (see graph to the right). The percentage of first cycle approvals of priority applications remained about the same: 57 percent in FY 2003 versus 55 percent in FY 2004.
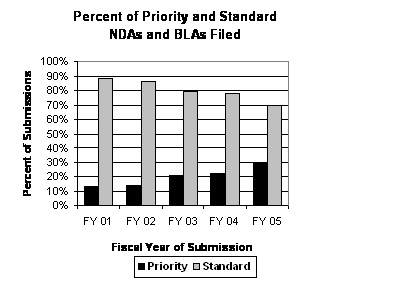
More Priority Applications Filed and Approved Under PDUFA III. The number of priority applications has steadily risen over the past five years and represent a larger workload for FDA reviewers. In FY 2001, priority applications represented 13 percent (13 of 104) of all original applications filed. While the total number of original applications increased over the next 5 years by 9 percent (from 104 in FY 2001 to 113 in FY 2005), the total number of priority applications steadily increased from 13 in FY 2001 to 34 in FY 2005. As a result, priority applications represented 30 percent (34 of 113) of all original applications filed in FY 2005 (see graph above). Despite these changes, the median time for approval of these applications has fallen (estimated at around 6 months in FY 2003 and FY 2004 with the most recent data available), and the percentage of priority applications being approved in the first cycle has remained above 50 percent in FY 2003 and FY 2004. Concurrently, the number of priority applications approved in 6 months or less has steadily increased from 2 in FY 2001 to 14 in FY 2004.
Workload Variations Under PDUFA III. FDA has seen significant variations to its workload under PDUFA III (FY 2003 through FY 2005). Almost all filings and submissions first increased, and then decreased. Concurrently, FDA reviewers faced significant increases in their workloads with respect to procedural and processing goals. FDA reviewers do not have prior knowledge of the workload each year and must adjust to the products that they receive.
| Selected Workload Under PDUFA III | |||
|---|---|---|---|
| Product/Request | FY 2003 | FY 2004 | FY 2005 |
| Original NDAs and BLAs | 109 | 129 | 113 |
| Priority NDAs and BLAs | 23 | 29 | 34 |
| Resubmitted NDAs and BLAs | 74 | 85 | 54 |
| NDA and BLA Efficacy Supplements | 153 | 204 | 153 |
| NDA and BLA Manufacturing Supplements | 2,598 | 2,500 | 2,503 |
| Meeting Requests | 2,119 | 2,284 | 2,430 |
| Special Protocol Assessments | 293 | 346 | 392 |
The tables below summarize FDA's review performance on the FY 2004 application submissions and the preliminary performance in reviewing FY 2005 application submissions and meeting other performance goals.
This section updates FDA's review performance on the FY 2004 application submissions and evaluates FDA's performance in reviewing FY 2005 application submissions and meeting other PDUFA performance goals. The following information refers to FDA performance presented in this section.
The table below summarizes the annual review time goals for original NDAs and BLAs. Over the 5-year period defined by PDUFA III, the goal of reviewing 90 percent of priority applications within 6 months and standard applications within 10 months remains constant.
| Original Application Type | Review Time Goal | Performance Goal FY 2003 – FY 2007 Submissions |
|---|---|---|
| Priority | 6 months | 90% on time |
| Standard | 10 months | 90% on time |
The total number of original applications in FY 2005 was lower than the FY 2004 level, but consistent with the trend of modest growth since FY 2001. During this same 5-year period, the number of priority applications increased each year from 13 in FY 2001 to 34 in FY 2005 when they represented almost one-third of the applications filed (see graph and table below).
| Original Applications Filed (Priority / Standard) |
|||||
|---|---|---|---|---|---|
| Type | FY 01 | FY 02 | FY 03 | FY 04 | FY 05 1 |
| NDAs | 96 (10/86) |
96 (12/84) |
101 (19/82) |
120 (26/94) |
104 (28/76) |
| BLAs | 8 (3/5) |
9 (3/6) |
8 (4/4) |
9 (3/6) |
9 (6/3) |
| PDUFA Total | 104 (13/91) |
105 (15/90) |
109 (23/86) |
129 (29/100) | 113 (34/79) |
| NMEs 2 | 32 (8/24) |
22 (8/14) |
29 (12/17) |
31 (16/15) |
30 (15/15) |
The 90 percent on-time review performance goal was exceeded for all priority and standard NDAs, NMEs, and BLAs in FY 2004. FDA reviewed and acted on all but one (28 of 29) priority application within 6 months. FDA reviewed and acted on all but three (97 of 100) standard applications within ten months (see table below).
| Original Application Type | Review Within | Type | Reviewed and Acted On | Number on Time | Percent on Time | PDUFA Performance Goal |
|---|---|---|---|---|---|---|
| Priority | 6 months |
All Applications | 29 | 28 | 97% | 90% |
| NMEs & BLAs | 19 | 19 | 100% | 90% | ||
| Standard | 10 months |
All Applications | 100 | 97 | 97% | 90% |
| NMEs & BLAs | 21 | 21 | 100% | 90% |
As of September 30, 2005 , over half (19 of 34) of the priority applications filed in FY 2005 had been reviewed and acted on; and all but one met the 6-month review performance goal. Approximately one-tenth (7 of 79) of the standard applications received had been reviewed and acted on; and all met the 10-month review performance goal (see table below). With submissions still pending and not overdue, it is too early to make a final performance determination for FY 2005.
| Original Application Type | Review Within | Type | Reviewed and Acted On | Number on Time | Percent on Time | PDUFA Performance Goal |
|---|---|---|---|---|---|---|
| Priority | 6 months |
All Applications | 19 | 18 | 95% | 90% |
| NMEs & BLAs | 13 | 12 | 92% | 90% | ||
| Standard | 10 months |
All Applications | 7 | 7 | 100% | 90% |
| NMEs & BLAs | 1 | 1 | 100% | 90% |
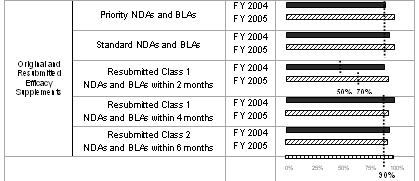
The table below summarizes the annual review time goals for resubmitted NDA and BLA applications. A resubmission is a firm's response after an FDA action of "approvable," "not approvable," or "complete response" on an application. The applicable performance goal for a resubmission is determined by the year in which the resubmission itself is received, rather than the year in which the original application was submitted. 3 Over the 5-year period defined by PDUFA III, the goal of reviewing 90 percent of Class 1 resubmitted applications within two months and Class 2 resubmitted applications within 6 months remains constant.
| Resubmitted Application Type | Review Time Goal | Performance Goal FY 2003 – FY 2007 Submissions |
|---|---|---|
| Class 1 | 2 months | 90% on time |
| Class 2 | 6 months | 90% on time |
The total number of resubmitted applications decreased in FY 2005. The reduction in FY 2005, however, was primarily in Class 2 resubmitted applications that decreased by almost half (see graph and table below).
| Resubmitted Applications (Class 1 / Class 2) |
|||||
|---|---|---|---|---|---|
| Type | FY 01 | FY 02 | FY 03 | FY 04 | FY 05 |
| NDAs | 62 (25/37) |
62 (20/42) |
62 (24/38) |
83 (21/62) |
51 (20/31) |
| BLAs | 16 (6/10) |
15 (2/13) |
12 (1/11) |
2 (1/1) |
3 (0/3) |
| PDUFA Total | 78 (31/47) |
77 (22/55) |
74 (25/49) |
85 (22/63) |
54 (20/34) |
The 90 percent on-time review performance goal was exceeded for both Class 1 and Class 2 resubmissions in FY 2004. FDA reviewed and acted on all Class 1 resubmitted applications within 2 months. FDA reviewed and acted on all but one (62 of 63) Class 2 resubmitted application within 6 months (see table below).
| Resubmitted Application Type | Review Within | Reviewed and Acted On | Number on Time | Percent on Time | PDUFA Performance Goal |
|---|---|---|---|---|---|
| Class 1 | 2 months | 22 | 22 | 100% | 90% |
| Class 2 | 6 months | 63 | 62 | 98% | 90% |
Of the 20 Class 1 resubmissions received in FY 2005, all but two met the 6-month review goal. As of September 30, 2005, almost half (16 of 34) of the Class 2 resubmissions received had been reviewed and acted on; and all but two met the 6-month review performance goal (see table below). With Class 2 resubmissions still pending and not overdue, it is too early to make a final performance determination for FY 2005.
| Resubmitted Application Type | Review Within | Reviewed and Acted On | Number on Time | Percent on Time | PDUFA Performance Goal |
|---|---|---|---|---|---|
| Class 1 | 2 months | 20 | 18 | 90% | 90% |
| Class 2 | 6 months | 16 | 14 | 88% | 90% |
The table below summarizes the annual review time goals for original efficacy supplements to NDAs and BLAs. Over the 5-year period defined by PDUFA III, the goal of reviewing 90 percent of priority supplements within 6 months and standard supplements within 10 months remains constant.
| Efficacy Supplement Type | Review Time Goal | Performance Goal FY 2003 – FY 2007 Submissions |
|---|---|---|
| Priority | 6 months | 90% on time |
| Standard | 10 months | 90% on time |
The number of efficacy supplements received in FY 2005 declined from the level in FY 2004, returning to the FY 2003 level. However, over recent years, the number of BLA efficacy supplements has increased with the number in FY 2005 almost three times higher than in FY 2002 (see graph and table below).
| Efficacy Supplements Filed
(Priority / Standard ) |
|||||
|---|---|---|---|---|---|
| Type | FY 01 | FY 02 | FY 03 | FY 04 | FY 05 |
| NDAs | 154 (7/147) |
159 (31/128) |
138 (35/103) |
183 (48/135) |
122 (23/99) |
| BLAs | 16 (2/14) |
11 (4/7) |
15 (2/13) |
21 (2/19) |
31 (5/26) |
| PDUFA Total | 170 (9/161) |
170 (35/135) |
153 (37/116) |
204 (50/154) |
153 (28/125) |
The 90 percent on-time review performance goal was exceeded for both priority and standard efficacy supplements in FY 2004. FDA reviewed and acted on all but four (46 of 50) priority efficacy supplements within 6 months. FDA reviewed and acted on all but six (148 of 154) standard efficacy supplements within 10 months (see table below).
| Efficacy Supplement Type | Review Within | Reviewed and Acted On | Number on Time | Percent on Time | PDUFA Performance Goal |
|---|---|---|---|---|---|
| Priority | 6 months | 50 | 46 | 92% | 90% |
| Standard | 10 months | 154 | 148 | 96% | 90% |
As of September 30, 2005 , almost two-thirds (18 of 28) of the priority efficacy supplements filed in FY 2005 had been reviewed and acted on; and all met the 6-month review performance goal. Almost one-fifth (23 of 125) of the standard efficacy supplements received had been reviewed and acted on; and all met the 10-month review performance goal (see table below). With submissions still pending and not overdue, it is too early to make a final performance determination for FY 2005.
| Efficacy Supplement Type | Review Within | Reviewed and Acted On | Number on Time | Percent on Time | PDUFA Performance Goal |
|---|---|---|---|---|---|
| Priority | 6 months | 18 | 18 | 100% | 90% |
| Standard | 10 months | 23 | 23 | 100% | 90% |
The table below summarizes the annual review time goals for resubmitted efficacy supplements to NDAs and BLAs. This is the third year for this goal under PDUFA III. For Class 1 resubmissions, the goal progresses from reviewing 30 percent of FY 2003 resubmissions in 2 months to 90 percent by FY 2007. Over the 5-year period defined by PDUFA III, the goal of reviewing 90 percent of Class 2 resubmissions within 6 months remains constant.
| Resubmitted Efficacy Supplement Type |
Review Time Goal |
Performance Goal | ||||
|---|---|---|---|---|---|---|
| FY 03 | FY 04 | FY 05 | FY 06 | FY 07 | ||
| Class 1 | 2 months | 30% | 50% | 70% | 80% | 90% |
| 4 months | -- | 90% | -- | |||
| 6 months | 90% | -- | ||||
| Class 2 | 6 months | 90% | ||||
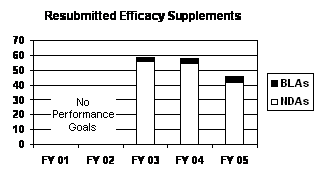
The total number of resubmitted efficacy supplements received in FY 2005 decreased by about one-fifth when compared to FY 2003 and FY 2004 levels. All of the decrease was represented by NDA supplements (see graph and table below).
| Resubmitted Efficacy Supplements (Class 1 / Class 2) | |||||
|---|---|---|---|---|---|
| Type | FY 01 4 | FY 02 4 | FY 03 | FY 04 | FY 05 |
| NDAs | -- | -- | 56 (16/40) | 55 (32/23) | 42 (23/19) |
| BLAs | -- | -- | 3 (1/2) | 3 (3/0) | 4 (1/3) |
| PDUFA Total | -- | -- | 59 (17/42) | 58 (35/23) | 46 (24/22) |
The on-time review performance goals were exceeded for both Class 1 and Class 2 efficacy supplement resubmissions in FY 2004. FDA reviewed and acted on all but three (32 of 35) Class 1 resubmitted efficacy supplements within the 2-month review performance goal; and all within the 4-month review performance goal. FDA reviewed and acted on all but one (22 of 23) Class 2 resubmitted efficacy supplement within the 6-month review performance goal (see table below).
| Resubmitted Efficacy Supplement Type | Review Within | Reviewed and Acted On | Number on Time | Percent on Time | PDUFA Performance Goal |
|---|---|---|---|---|---|
| Class 1 | 2 months | 35 | 32 | 91% | 50% |
| 4 months | 35 | 100% | 90% | ||
| Class 2 | 6 months | 23 | 22 | 96% | 90% |
As of September 30, 2005, most (19 of 24) of the Class 1 resubmitted efficacy supplements had been reviewed and acted on; and all but 1 met the 2-month review performance goal and the 4-month review performance goal. Most (18 of 22) of the Class 2 resubmitted efficacy supplements had been reviewed and acted on; and all but 1 met the 6-month review performance goal (see table below). With resubmissions still pending and not overdue, it is too early to make a final performance determination for FY 2005.
| Resubmitted Efficacy Supplement Type | Review Within | Reviewed and Acted On | Number on Time | Percent on Time | PDUFA Performance Goal |
|---|---|---|---|---|---|
| Class 1 | 2 months | 19 | 18 | 95% | 70% |
| 4 months | 18 | 95% | 90% | ||
| Class 2 | 6 months | 18 | 17 | 94% | 90% |
The table below summarizes the annual review time goals for manufacturing supplements to NDAs and BLAs. Over the 5-year period defined by PDUFA III, the performance goal for manufacturing supplements that require FDA's approval before the changes can be enacted is 90 percent of supplements within 4 months of submission. The PDUFA performance goal for manufacturing supplements that do not require FDA's approval before the changes can be enacted is 90 percent of supplements within 6 months of submission. The manufacturing supplement goals remain constant.
| Manufacturing Supplement Type | Review Time Goal | Performance Goal FY 2003 – FY 2007 Submissions |
|---|---|---|
| Prior Approval Required | 4 months | 90% on time |
| Prior Approval Not Required | 6 months | 90% on time |
The total number of manufacturing supplements filed has been relatively steady over the past 4 years (FY 2002 through FY 2005) following a significant increase after FY 2001, with virtually no change between FY 2004 and FY 2005 levels. BLA supplements, which decreased slightly in FY 2005, continue to represent about one-third of all manufacturing supplements (see graph and table below).
| Manufacturing Supplements Filed (Prior Approval / No Prior Approval) | |||||
|---|---|---|---|---|---|
| Type | FY 01 | FY 02 | FY 03 | FY 04 | FY 05 |
| NDAs | 1,474 (579/895) |
1,759 (602/1,157) |
1,696 (618/1,078) |
1,617 (524/1,093) |
1,668 (645/1,023) |
| BLAs | 591 (185/406) |
717 (228/489) |
902
(303/599) |
883 (299/584) |
835 (259/576) |
| PDUFA Total | 2,065 (764/1,301) |
2,476 (830/1,646) |
2,598 (921/1,677) |
2,500 (823/1,677) |
2,503 (904/1,599) |
The 90 percent on-time review performance goal was exceeded for both types of manufacturing supplements in FY 2004. FDA reviewed and acted on all 823 manufacturing supplements that required prior approval and all 1,677 manufacturing supplements that did not require prior approval. Ninety-six percent (789 of 823) met the 4-month review performance goal. Ninety-nine percent (1,656 of 1,677) met the 6-month review performance goal (see table below).
| Manufacturing Supplement Type | Review Within | Reviewed and Acted On | Number on Time | Percent on Time | PDUFA Performance Goal |
|---|---|---|---|---|---|
| Prior Approval Required | 4 months | 823 | 789 | 96% | 90% |
| Prior Approval Not Required | 6 months | 1,677 | 1,656 | 99% | 90% |
As of September 30, 2005, over two-thirds (638 of 904) of the manufacturing supplements requiring prior approval had been reviewed and acted on; and 97 percent (617 of 638) were reviewed within the 4-month review performance goal. Over one-half (985 of 1,599) of the manufacturing supplements not requiring prior approval had been reviewed and acted on; and 99 percent (976 of 985) were reviewed within the 6-month review performance goal (see table below). With submissions still pending and not overdue, it is too early to make a final performance determination for FY 2005.
| Manufacturing Supplement Type | Review Within | Reviewed and Acted On | Number on Time | Percent on Time | PDUFA Performance Goal |
|---|---|---|---|---|---|
| Prior Approval Required | 4 months | 638 | 617 | 97% | 90% |
| Prior Approval Not Required | 6 months | 985 | 976 | 99% | 90% |
This section presents FDA's performance in achieving the FY 2005 procedural and processing goals and accomplishments for PDUFA III initiatives and commitments. The following information refers to FDA performance presented in this section.
The procedural and processing goals FDA committed to achieve were designed to improve application submissions and FDA-sponsor interactions during new drug development and application review. The table below summarizes the meeting management goals that address meeting requests, scheduling meetings, and preparing meeting minutes.
| Action | Performance Goal | Performance Level FY 2003 – FY 2007 |
|---|---|---|
| Meeting Requests | Notify requestor of formal meeting in writing within 14 days of request. | 90% on time |
| Scheduling Meetings | Schedule meetings within goal date (within 30 days of receipt of request for Type A meetings, 60 days for Type B meetings, and 75 days for Type C meetings). If the requested date for any of these types of meetings is greater than 30, 60, or 75 days, as appropriate, from the date the request is received by FDA, the meeting date should be within 14 days of the requested date. | 90% on time |
| Meeting Minutes | FDA-prepared minutes, clearly outlining agreements, disagreements, issues for further discussion, and action items will be available to the sponsor within 30 days of meeting. | 90% on time |
The number of meeting requests and, subsequently, meetings scheduled, both increased for the fifth straight year, reaching all time highs (see graph and table below).
| Meeting Management | |||||
|---|---|---|---|---|---|
| Type | FY 01 | FY 02 | FY 03 | FY 04 | FY 05 |
| Meeting Request Notifications | 1,662 | 1,745 | 2,119 | 2,284 | 2,430 |
| Scheduling Meetings | 1,546 | 1,643 | 2,002 | 2,125 | 2,193 |
| Meeting Minutes | 1,395 | 1,503 | 1,761 | 1,854 | 1,905 |
As of September 30, 2005 , FDA had responded to almost all (2,379 of the 2,430) meeting requests received in FY 2005, over three-fourths (2,028 of 2,193) of the Type A, Type B, and Type C meetings had been scheduled, and approximately two-thirds (1,513 of 1,905) of the meeting minutes had been completed. With meeting requests, scheduling, and minutes still pending within goal, it is too early to make a final performance determination for FY 2005. Preliminary performance regarding meeting management indicated:
Meeting Requests. Eighty-nine percent (2,123 of 2,379) of meeting requests were responded to within goal.
Scheduling Meetings. Eighty-eight percent (1,780 of 2,028) of Type A, Type B, and Type C meetings were scheduled within goal.
Meeting Minutes. Eighty-three percent (1,249 of 1,513) of meeting minutes had been completed.
| Meeting Requests/Scheduling/Mintues | CBER | Total | Met Goal | Missed Goal 5 | Pending Within Goal | Percent on Time 6 | PDUFA Performance Goal | |
|---|---|---|---|---|---|---|---|---|
| Meeting Requests | CBER | 256 | 251 | 5 | 0 | |||
| CDER | 2,174 | 1,872 | 251 | 51 | ||||
| Combined | 2,430 | 2,123 | 256 | 51 | 89% | 90% | ||
| Scheduling Meetings | Type A | CBER | 8 | 6 | 0 | 2 | ||
| CDER | 351 | 151 | 101 | 99 | ||||
| Type B | CBER | 137 | 114 | 3 | 20 | |||
| CDER | 1,046 | 908 | 112 | 26 | ||||
| Type C | CBER | 79 | 76 | 1 | 2 | |||
| CDER | 572 | 525 | 31 | 16 | ||||
| All | CBER | 224 | 196 | 4 | 24 | |||
| CDER | 1,969 | 1,584 | 244 | 141 | ||||
| Combined | 2,193 | 1,780 | 248 | 165 | 88% | 90% | ||
| Meeting Minutes | CBER | 164 | 150 | 6 | 8 | |||
| CDER | 1,741 | 1,099 | 258 | 384 | ||||
| Combined | 1,905 | 1,249 | 264 | 392 | 83% | 90% | ||
The table below summarizes the annual performance goal for the response to the requests for special protocol assessments. Over the 5-year period defined by PDUFA III, the goal of responding to 90 percent of sponsor's request for evaluation of protocol design within 45 days of receipt remains constant.
| Action | Performance Goal | Performance Level FY 2003 – FY 2007 |
|---|---|---|
| Special Protocol Question Assessment and Agreement | Respond to sponsor's request for evaluation of protocol design within 45 days of receipt. | 90% on time |
Special protocol assessments increased for the fifth straight year in FY 2005, reaching an all time high of more than triple the number in FY 2001 (see graph and table below).
| FY 01 | FY 02 | FY 03 | FY 04 | FY 05 |
|---|---|---|---|---|
| 125 | 248 | 293 | 346 | 392 |
As of September 30, 2005 , FDA had responded to most (350 of 392) of the sponsors' requests for evaluation of protocol designs received in FY 2005. FDA did not meet the performance goal for response time to requests for special protocol assessments. The preliminary data indicates that FDA is just short (88 percent) of the performance goal. There are 42 protocol assessments pending within goal; however, responses to these requests for special protocol assessments will not enable FDA to meet the FY 2005 performance goal.
| Total | Met Goal | Missed Goal | Pending Within Goal | Percent on Time | PDUFA Performance Goal |
|---|---|---|---|---|---|
| 392 (8/384) |
307 (8/299) |
43 (0/43) |
42 (0/42) |
88% | 90% |
The table below summarizes the annual performance goal for the response to clinical holds. Over the 5-year period defined by PDUFA III, the goal of responding to sponsor's complete response to a clinical hold within 30 days of receipt remains constant.
| Action | Performance Goal | Performance Level FY 2003 – FY 2007 |
|---|---|---|
| Response to Clinical Hold | Respond to sponsor's complete response to a clinical hold within 30 days of receipt. | 90% on time |
Responses to clinical holds have been relatively level over the past 3 years, FY 2003 through FY 2005 (see graph and table below).
| FY 01 | FY 02 | FY 03 | FY 04 | FY 05 |
|---|---|---|---|---|
| 158 | 171 | 136 | 135 | 129 |
As of September 30, 2005 , FDA had responded to almost all (118 of 129) of sponsors' complete responses to clinical holds received in FY 2005. FDA did not meet the performance goal for response time to clinical holds. The preliminary data show that 83 percent were responded to within goal. There are 11 clinical holds pending within goal; however, the responses to these holds will not enable FDA to meet the FY 2005 performance goal.
| Total | Met Goal | Missed Goal | Pending Within Goal | Percent on Time | PDUFA Performance Goal |
|---|---|---|---|---|---|
| 129 (42/87) |
98 (36/62) |
20 (1/19) |
11 (5/6) |
83% | 90% |
The table below summarizes the annual performance goal for the response to major dispute resolutions. Over the 5-year period defined by PDUFA III, the goal of responding to sponsor's appeal of decision within 30 days of receipt remains constant.
| Action | Performance Goal | Performance Level FY 2003 – FY 2007 |
|---|---|---|
| Major Dispute Resolution | Respond to sponsor's appeal of decision within 30 days of receipt. | 90% on time |
Major dispute resolutions remained relatively steady across the 5-year period with the exception of the FY 2003 level (see graph and table below).
| FY 01 | FY 02 | FY 03 | FY 04 | FY 05 |
|---|---|---|---|---|
| 11 | 12 | 20 | 10 | 9 |
As of September 30, 2005 , FDA had responded to all (9 of 9) sponsors' appeals of decisions received in FY 2005. FDA missed the response time for one major dispute resolution, resulting in a performance level one percent under the goal.
| Total | Met Goal | Missed Goal | Pending Within Goal | Percent on Time | PDUFA Performance Goal |
|---|---|---|---|---|---|
| 9 (2/7) |
8 (2/6) |
1 (0/1) |
0 (0/0) |
89% | 90% |
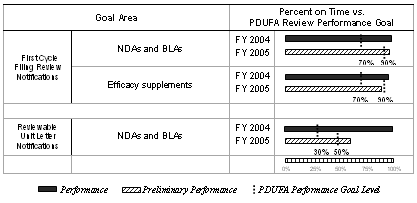
The table below summarizes the annual review time goals for first cycle filing review notifications for original NDAs and BLAs, and efficacy supplements. This is the third year for this goal under PDUFA III. FDA is to report substantive deficiencies (or lack of same) identified during the initial filing review to the sponsor by letter, telephone conference, facsimile, secure e-mail, or other expedient means within 14 days after the 60-day filing date. Performance levels progress from 50 percent on time for FY 2003 submissions to 90 percent for FY 2005 to FY 2007 submissions.
| First Cycle Filing Review Notification Type | Review Time Goal | Performance Level | ||||||
|---|---|---|---|---|---|---|---|---|
| FY 03 | FY 04 | FY 05 | FY 06 | FY 07 | ||||
| Original NDAs and BLAs | Within 14 days after 60-day filing date | 50% | 70% | 90% | 90% | 90% | ||
| Efficacy Supplements | Within 14 days after 60-day filing date | 50% | 70% | 90% | 90% | 90% | ||
The total number of FY 2005 first cycle filings decreased by 17 percent from the level in FY 2004, returning to the FY 2003 levels (see graph and table below).
| Type | FY 01 4 | FY 02 4 | FY 03 | FY 04 | FY 05 |
|---|---|---|---|---|---|
| NDAs | -- | -- | 104 | 123 | 104 |
| BLAs | -- | -- | 8 | 9 | 9 |
| Total | -- | -- | 112 | 132 | 113 |
| Efficacy Supplements 7 | -- | -- | 121 | 147 | 118 |
The on-time review performance goals were exceeded for all first cycle filing review notifications in FY 2004. FDA completed initial filing reviews for all but 3 (129 of 132) original NDAs and BLAs within 14 days after the 60-day filing date. FDA completed initial filing reviews for all but 6 (141 of 147) efficacy supplements within 14 days after the 60-day filing date (see table below).
| First Cycle Filing Review Notification Type | Review Within | Initial Filing Reviews | Number on Time | Percent on Time | PDUFA Performance Goal |
|---|---|---|---|---|---|
| NDAs and BLAs | Within 14 days after 60-day filing date | 132 | 129 | 98% | 70% |
| Efficacy Supplements | Within 14 days after 60-day filing date | 147 | 141 | 96% | 70% |
As of September 30, 2005 , three-fourths (84 of 113) of NDAs and BLAs had received an initial filing review; and 96 percent (81 of 84) were reviewed within 14 days after the 60-day filing date. Most (101 of 118) efficacy supplements had been reviewed; and 89 percent (90 of 101) were reviewed within goal (see table below). With submissions still pending and not overdue, it is too early to make a final performance determination for FY 2005.
| First Cycle Filing Review Notification Type | Review Within | Initial Filing Reviews | Number on Time | Percent on Time | PDUFA Performance Goal |
|---|---|---|---|---|---|
| NDAs and BLAs | Within 14 days after 60-day filing date | 84 | 81 | 96% | 90% |
| Efficacy Supplements | Within 14 days after 60-day filing date | 101 | 90 | 89% | 90% |
The table below summarizes the annual review time goals for reviewable unit letter notifications for NDAs and BLAs. This is the second year for this goal under PDUFA III. Under the Continuous Marketing Application Pilot 1 program, applicants may submit a portion of their marketing application, reviewable unit (RU), before submitting the complete application for Fast Track Original NDAs and BLAs, based on meeting specific criteria for inclusion in the Pilot. An NDA or BLA may have more than one RU. Each RU is tracked independently. Under this goal, FDA is to issue discipline review letters for pre-submitted RUs to NDAs and BLAs within 6 months of receipt. Performance levels progress from 30 percent on time for FY 2004 submissions to 90 percent for FY 2007 submissions.
| Reviewable Unit Type | Review Time Goal | Performance Level | ||||
|---|---|---|---|---|---|---|
| FY 03 | FY 04 | FY 05 | FY 06 | FY 07 | ||
| NDA | 6 months | -- | 30% | 50% | 70% | 90% |
| BLA | 6 months | -- | 30% | 50% | 70% | 90% |
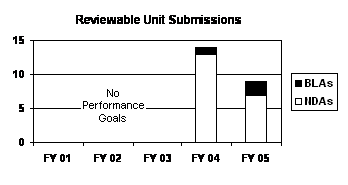
The total number of NDA reviewable unit submissions decreased from FY 2004 to FY 2005 (see graph and table below).
| Type | FY 01 4 | FY 02 4 | FY 03 4 | FY 04 | FY 05 |
|---|---|---|---|---|---|
| NDAs | -- | -- | -- | 13 | 7 |
| BLAs | -- | -- | -- | 1 | 2 |
| PDUFA Total | -- | -- | -- | 14 | 9 |
FDA performance on all reviewable unit letter notifications exceeded the 30 percent on-time review performance goal in FY 2004. FDA reviewed and acted on all reviewable unit submissions within 6 months (see table below).
| Reviewable Unit Type | Review Within | Reviewed and Acted On | Number on Time | Percent on Time | PDUFA Performance Goal |
|---|---|---|---|---|---|
| NDAs and BLAs | 6 months | 14 | 14 | 100% | 30% |
As of September 30, 2005 , over one-half (5 of 9) of NDA and BLA reviewable unit submissions had been reviewed and acted on; and most (3 of 5) were reviewed within the 6-month review time goal (see table below). With reviewable unit submissions still pending and not overdue, it is too early to make a final performance determination for FY 2005.
| Reviewable Unit Type | Review Within | Reviewed and Acted On | Number on Time | Percent on Time | PDUFA Performance Goal |
|---|---|---|---|---|---|
| NDAs and BLAs | 6 months | 5 | 3 | 60% | 50% |
The management initiatives FDA committed to achieve under PDUFA III were designed to improve the overall application review process.
The first Continuous Marketing Application (CMA) pilot (Pilot 1) applies to fast track products that have demonstrated significant promise as a therapeutic advance in clinical trials, and will provide an early discipline review of the reviewable units (RUs) of the sponsor's NDA/BLA submitted in advance of the complete application. (The CMA Pilot 1 program became effective when the final guidance was published on October 6, 2003 , and is available at http://www.fda.gov/cder/guidance/5739-fnl.pdf .)
The second CMA pilot (Pilot 2) also applies to fast track products and provides for FDA-sponsor agreement to engage in frequent scientific feedback and interactions during the clinical trial phase of product development. (The CMA Pilot 2 program became effective when the final guidance was published on October 6, 2003 , and is available at http://www.fda.gov/cder/guidance/5740-fnl.pdf .)
FY 2005 Accomplishments : As of September 30, 2005 , a cumulative total of 12 products had been identified for inclusion in the Pilot 1 program. Nine RUs were received during FY 2005. As of September 30, 2005 , 56 percent (5 of 9) of the RUs received had been reviewed and acted on; and 60 percent (3 of 5) were within the goal time. Additionally, a total of nine products were participating in the Pilot 2 program as of September 30, 2005 . In August 2005, FDA awarded a task order under an existing contract to evaluate the CMA Pilots.
Approvals that take more than one review cycle to complete are generally not in the best interest of the public, the FDA, or the sponsor submitting the product application. Although additional review cycles are sometimes necessary to resolve important issues regarding safety, quality, or efficacy; in most cases, the extra cycles could be avoided, saving time and effort. For applications that are ultimately approved, the causes of multiple review cycles can include deficiencies in sponsors' applications, communication problems during the review process, or difficulty achieving final resolution on such topics as labeling.
Efforts to improve the first cycle review process include an initiative for notification of substantive deficiencies identified during the initial filing review for original NDAs and BLAs and an initiative to develop and publish Good Review Management Principles (GRMP) with provisions for both FDA reviewers and industry sponsors. The notification initiative was implemented on October 2, 2002 .
FY 2005 Accomplishments : As of September 30, 2005 , 74 percent (84 of 113) of NDAs and BLAs and 86 percent (101 of 118) of efficacy supplements had received an initial filing review. 7 In January 2005, FDA awarded a task order under an existing contract to conduct a retrospective analysis of first cycle reviews. The final GRMP guidance was published on March 31, 2005 , and is available at http://www.fda.gov/cder/guidance/5812fnl.htm . A joint CDER and CBER rollout of the GRMP guidance took place in April 2005.
Under the PDUFA III performance management goal, FDA will conduct initiatives that are targeted to improve the new drug review process. FDA will also contract with outside expert consultants for analysis, training, and technical assistance to help implement a quality systems approach to the new drug review process.
FY 2005 Accomplishments: In November 2004, FDA established a Quality Systems Group to coordinate the implementation of a quality management system for new drug review and PDUFA III performance management initiatives. Contracts have been awarded for such projects as process improvements in CDER's Office of New Drugs and Office of Drug Safety, quality meeting minutes, leadership development and quality systems training, and managerial costing.
This PDUFA III initiative allows a sponsor to request that FDA engage an independent expert consultant during the development period for certain biotechnology products. The consultant would be selected by FDA to assist in the FDA's review of the protocol for the clinical studies that would support the claims for the product. This initiative is intended to facilitate product development. Final guidance was published on August 18, 2004 , and is available at: http://www.fda.gov/cber/gdlns/bioclin.htm .
FY 2005 Accomplishments : So far no sponsors have requested assistance under the program.
The initiative to address postmarket risk before an application is submitted, during the review process, and during the peri-approval period (two or three years post-approval) will facilitate postmarket risk management by helping FDA better understand any risks and by providing feedback to the sponsors. Guidances will be published for three areas: Good Risk Assessment, Risk Management, and Pharmacovigilance Practices.
FY 2005 Accomplishments : FDA met the PDUFA III goal for CDER and CBER to jointly develop final guidance documents that address good risk assessment, risk management, and pharmacovigilance practices by the publication in the Federal Register on March 29, 2005 , of the following Guidances for Industry:
• Premarketing Risk Assessment http://www.fda.gov/cder/guidance/6357fnl.pdf
• Development and Use of Risk Minimization Action Plans http://www.fda.gov/cder/guidance/6358fnl.pdf
• Good Pharmacovigilance Practices and Pharmacoepidemiologic Assessment http://www.fda.gov/cder/guidance/6359OCC.pdf
CDER and CBER have provided training to their staff on these three guidance documents. Additionally , FDA reviewed 34 Risk Management Plans (RMPs) of which 12 were for PDUFA III products. FDA also participated in 38 PDUFA III pre-NDA/BLA review meetings, 23 PDUFA III pre-approval safety conferences, 3 PDUFA III peri-approval RMP reviews, and the evaluation of 7 active RMPs for non-PDUFA III products.
The electronic applications and submissions commitments under PDUFA III were designed to improve the overall application review process.
Centralize the accountability and funding for all PDUFA IT initiatives/activities under the leadership of the FDA Chief Information Officer (CIO).
FY 2005 Accomplishments: In FY 2005, the FDA implemented "earned value management" for major IT investments to provide a single view of cost and performance of a project for use in project stage gate reviews and reporting to the CIO, HHS, and the Office of Management and Budget (OMB). As part of the earned value management implementation, the FDA has established standard contract language for all IT investments and is working with FDA Office of Acquisitions and Grants Services to implement this language in new and reissued FDA IT contracts.
Periodically review and evaluate the progress of IT initiatives against project milestones. This includes, on an annual basis, an assessment of progress against PDUFA III IT goals and established program milestones, including appropriate changes to plans.
FY 2005 Accomplishments : This FY 2005 PDUFA Performance Report to the President and the Congress satisfies the annual requirement. In addition, the FDA reported IT progress to stakeholders at the PDUFA IT quarterly briefings (December 2004, February 2005, May 2005, and September 2005) and through PhRMA/BIO PDUFA updates (February 2005 and May 2005).
Implement a common solution for the secure exchange of application content.
FY 2005 Accomplishments : The FDA has continued to participate and provide guidance on the Secure Access For Everyone (SAFE) standard for the biopharmaceutical industry. Over the last two years, the FDA has been performing an advisory role on the SAFE initiative that is designed for the purpose of simplifying, securing, and streamlining business-to-regulator information exchange. The SAFE standard consists of policies, procedures, guidelines, technical specifications, and a legal and liability risk management framework for ensuring the validity of the electronic signatures used for information exchange and electronic submissions to regulators.
Deliver a single point of entry for the receipt and processing of all electronic submissions in a highly secure environment.
FY 2005 Accomplishments : In January 2005, the FDA awarded the contract to develop the Electronic Submissions Gateway (ESG). Accomplishments include completion of the setup and internal (FDA) testing of the Gateway infrastructure and software. ESG is an FDA-wide solution for accepting electronic regulatory submissions. The ESG will provide a single point of entry for the secure submission of regulatory information to the FDA. The ESG will be a replacement of the current electronic gateway used for mandatory safety reporting by drug and biologic manufacturers and will provide a number of enhancements to drug safety reporting, including the acceptance of attachments and XML reporting.
Phase 1 of the FDA ESG will support the receipt of electronic regulatory submissions of up to 100GBs in size to CBER, CDER, and CDRH. The roll-out of Phase 1 will be preceded by a pilot testing program where volunteers from industry will use the FDA ESG to send test submissions and provide feedback on the new infrastructure.
Provide a format and review system for the electronic submission of the Common Technical Document (e-CTD).
FY 2005 Accomplishments : In the first quarter of FY 2005, the FDA performed an alternatives analysis to determine if the commercial market could meet FDA requirements for the review of e-CTD submissions. This analysis compared the FDA developed review tool, the e-CTD Viewer System (EVS), with a number of commercial products. The analysis concluded that it would be more efficient to acquire a commercial tool. Based on the alternatives analysis, the increasing number of e-CTD submissions, and working through the project and PDUFA governance process, the commercial software was implemented at CBER and CDER in the third quarter of FY 2005.
In FY 2005, over 1,000 e-CTD submissions were received by CBER and CDER, including over 800 submissions crossing over 79 marketing applications (NDA and BLA), and over 200 submissions crossing over 24 INDs. The e-CTD guidance and specifications are available at http://www.fda.gov/cder/regulatory/ersr/ectd.htm .
Conduct an objective analysis and develop a plan for consolidation of PDUFA III IT infrastructure.
FY 2005 Accomplishments : FDA continues to make progress in the consolidation of its IT infrastructure through collaboration with HHS in achieving its "One HHS" goals and objectives and ongoing efforts to accomplish the IT consolidation goals as part of PDUFA. To meet these goals and requirements, the CIO established an Enterprise Infrastructure Management framework to manage the various IT consolidation programs. These programs are the Enterprise Email System (EES), Enterprise Storage/Backup Management, Enterprise Systems Management, IT Consolidation, Capacity Management, and White Oak. FY 2005 accomplishments include:
• IT Infrastructure Service Contracts. Consolidating 15 IT Infrastructure Service contracts into a Single Source Infrastructure Service Support contract.
• White Oak Consolidated Telecommunication Infrastructure. The White Oak consolidated telecommunication infrastructure is up and operational. In September 2005, FDA staff began migrating 1,700 employees to the campus who will use the Voice over IP telephony system, where video, voice, and data are integrated into one system.
• Phased Server and Storage Consolidation. As part of the IT Consolidation program to modernize the FDA's infrastructure, the FDA has been finalizing strategies for the initiation of phased server and storage consolidation in FY 2007.
• Secure "One HHS". FDA continued with its efforts to identify, and benefit from, opportunities to consolidate with other HHS programs by successfully meeting the goals of the Secure "One HHS" program. Meeting these goals supported FDA efforts for infrastructure consolidation.
• HHS Enterprise Email System (EES). Another consolidation strategy has been unifying e-mail systems across HHS in order to take advantage of economies of scale and common standards.
Implement Capability Maturity Model (CMM) and include other industry best practices to ensure quality, efficiency, and cost effectiveness.
FY 2005 Accomplishments : In FY 2005, the FDA continued to strengthen and improve the FDA's IT project management capabilities. The Project Management Office further standardized systems development in the FDA and initiated project stage gate reviews to ensure conformance. The FDA also continued the project management certification training program. By the end of FY 2005, over 25 project managers had received their Project Management Professional Certification.
Use the same software applications where common business needs exist.
FY 2005 Accomplishments : In FY 2005, FDA developed a draft target architecture that will be the planning framework for future enhancements to the IT environment.
Develop a PDUFA III IT 5-year plan.
FY 2005 Accomplishments : An update to the March 2003 PDUFA IT Plan, that met the requirements of this performance goal, was completed in June 2004 and released at the September 2004 PDUFA IT quarterly briefing.
The table below summarizes, by fiscal year, the performance measures set forth in the letters referenced in the Food and Drug Administration Modernization Act of 1997 (PDUFA II) and in the Public Health Security and Bioterrorism Preparedness and Response Act of 2002 (PDUFA III). Goal summaries for the earlier years of PDUFA II can be found in the Appendix of earlier PDUFA Performance Reports. The complete text of the commitment letters is on the Internet at http://www.fda.gov/oc/pdufa/default.htm.
| Performance Goals | On-time Performance Level for Fiscal Year of Filing or Receipt | ||||||
|---|---|---|---|---|---|---|---|
| 2003 | 2004 | 2005 | 2006 | 2007 | |||
| Review and act on priority original NDAs and BLAs within 6 months of receipt. 8 | 90% | 90% | 90% | 90% | 90% | ||
| Review and act on standard original NDAs and BLAs within 10 months of receipt.8 | 90% | 90% | 90% | 90% | 90% | ||
| Review and act on priority efficacy supplements within 6 months of receipt. 8 | 90% | 90% | 90% | 90% | 90% | ||
| Review and act on standard efficacy supplements within 10 months of receipt. 8 | 90% | 90% | 90% | 90% | 90% | ||
| Review and act on all manufacturing supplements within 6 months of receipt and those requiring prior approval within 4 months of receipt.9 | 90% | 90% | 90% | 90% | 90% | ||
| Review and act on Class 1 resubmitted original applications within 2 months of receipt. | 90% | 90% | 90% | 90% | 90% | ||
| Review and act on Class 2 resubmitted original applications within 6 months of receipt. 8 | 90% | 90% | 90% | 90% | 90% | ||
| Review and act on Class 1 resubmitted efficacy supplements within | 2 months of receipt | 30% | 50% | 70% | 80% | 90% | |
| 4 months of receipt | -- | 90% | 90% | 90% | -- | ||
| 6 months of receipt | 90% | 90% | 90% | 90% | -- | ||
| Review and act on Class 2 resubmitted efficacy supplements within 6 months of receipt. 8 | 90% | 90% | 90% | 90% | 90% | ||
The performance goals for priority and standard original NMEs will be the same as for all of the original NDAs but will be reported separately. For biological products, for purposes of this performance goal, all original BLAs will be considered to be NMEs.
| Performance Area | FDA Activity | Performance Goal | Performance Level FY 2003 – FY 2007 |
|---|---|---|---|
| Meeting Management | Meeting Requests -- Notify requestor of formal meeting in writing (date, time, place, and participants). | Within 14 days of receipt of request. | 90% on time |
| Scheduling Meetings -- Schedule meetings within goal date or within 14 days of requested date if longer than goal date. | Type A Meetings within 30 days of receipt of request. | 90% on time | |
| Type B Meetings within 60 days of receipt of request. | 90% on time | ||
| Type C Meetings within 75 days of receipt of request. | 90% on time | ||
| Meeting Minutes -- FDA prepared minutes, clearly outlining agreements, disagreements, issues for further discussion and action times will be available to sponsor. | Within 30 days of meeting. | 90% on time | |
| Clinical Holds | Response to sponsor's complete response to a clinical hold. | Within 30 days of receipt of sponsor's response. | 90% on time |
| Special Protocol Question Assessment and Agreement | Response to sponsor's request for evaluation of protocol design. | Within 45 days of receipt of protocol and questions. | 90% on time |
| Major Dispute Resolution | Response to sponsor's appeal of decision. | Within 30 days of receipt of sponsor's appeal. | 90% on time |
| Performance Area | Initiative | Commitment | Performance Level and/or Implementation Timeline By Fiscal Year |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 2003 | 2004 | 2005 | 2006 | 2007 | |||||||
| Continuous Marketing Application | To test whether providing early review of selected applications and additional feedback and advice to sponsors during drug development for selected products can further shorten drug development and review times. | Discipline review team of a "reviewable unit" for a Fast Track drug or biologic will be completed and a DRL issued within 6 months of the date of the submission. | --- | 30% | 50% | 70% | 90% | ||||
| Independent Consultants for Biotechnology Clinical Trial Protocols | During the development period for a biotechnology product, a sponsor may request that FDA engage an independent expert consultant, selected by FDA, to participate in FDA's review of the protocol for the clinical studies that are expected to serve as the primary basis for a claim. | If FDA denies request, it must provide a written rationale within 14 days of receipt. | 100% | 100% | 100% | 100% | 100% | ||||
| First Cycle Review Performance Proposal | For original NDA/BLA applications and efficacy supplements, FDA will report substantive deficiencies (or lack of same) identified in the initial filing review to the sponsor by letter, telephone conference, facsimile, secure e-mail, or other expedient means. | FDA will provide the sponsor a notification of deficiencies (or lack of same) within 14 days after the 60-day filing date. | 50% | 70% | 90% | 90% | 90% | ||||
| Improving FDA Performance Management | Two specific initiatives will begin early in PDUFA III, supported from performance management initiative funds: 1) evaluation of first cycle review performance, and 2) process review and analysis within the two centers. | In FY 2003, FDA will contract with an outside consultant to conduct a comprehensive process review and analysis within CDER and CBER. | X | --- | --- | --- | --- | ||||
| Risk Management | Pre-NDA/BLA Meeting with Industry: The intent of these discussions will be for FDA to get a better understanding of the safety issues associated with the particular drug/biologic and the proposed risk management plans, and to provide industry with feedback on these proposals so that they can be included in the NDA/BLA submission. | By the end of FY 2004, CDER and CBER will jointly develop final guidance documents that address good risk assess-ment, risk management, and pharma-covigilance practices. | --- | X | --- | --- | --- | ||||
| -- | Not applicable | |||||||
| X | Action due |
| Initiatives | Implementation Deadline by Fiscal Year | ||||
|---|---|---|---|---|---|
| 2003 | 2004 | 2005 | 2006 | 2007 | |
| The Agency will centralize the accountability and funding for all PDUFA Information Technology initiatives/activities for CBER, CDER, ORA and OC under the leadership of the FDA CIO. The July 2001 HHS IT 5-year plan states that infrastructure consolidation across the department should be achieved, including standardization. The Agency CIO will be responsible for ensuring that all PDUFA III IT infrastructure and IT investments support the Agency's common IT goals, fit into a common computing environment, and follow good IT management practices. | X | X | X | X | X |
| The Agency CIO will chair quarterly briefings on PDUFA IT issues to periodically review and evaluate the progress of IT initiatives against project milestones, discuss alternatives when projects are not progressing, and review proposals for new initiatives. On an annual basis, an assessment will be conducted of progress against PDUFA III IT goals and, established program milestones, including appropriate changes to plans. A documented summary of the assessment will be drafted and forwarded to the Commissioner. A version of the study report redacted to remove confidential commercial or security information, or other information exempt from disclosure, will be made available to the public. The project milestones, assessment, and changes will be part of the annual PDUFA III report. | X | X | X | X | X |
| FDA will implement a common solution in CBER, CDER, ORA, and OC for the secure exchange of content, including secure e-mail, electronic signatures, and secure submission of, and access to, application components. | --- | --- | --- | --- | X |
| FDA will deliver a single point of entry for the receipt and processing of all electronic submissions in a highly secure environment. This will support CBER, CDER, OC, and ORA. The system should automate the current electronic submission processes such as checking the content of electronic submissions for completeness and electronically acknowledging submissions. | --- | --- | --- | --- | X |
| FDA will provide a specification format for the electronic submission of the Common Technical Document (e-CTD), and provide an electronic review system for this new format that will be used by CBER, CDER, and ORA reviewers. Implementation should include training to ensure successful deployment. This project will serve as the foundation for automation of other types of electronic submissions. The review software will be made available to the public. | --- | --- | --- | --- | X |
| Within the first 12 months, FDA will conduct an objective analysis and develop a plan for consolidation of PDUFA III IT infrastructure and desktop management services activities that will access and prioritize the consolidation possibilities among CBER, CDER, ORA, and OC to achieve technical efficiencies, target potential savings and realize cost efficiencies. Based upon the results of this analysis, to the extent appropriate, establish common IT infrastructure and architecture components according to specific milestones and dates. A documented summary of analysis will be forwarded to the Commissioner. A version of the study report, redacted to remove confidential commercial or security information, or other information exempt from disclosure, will be made available to the public. | --- | X | --- | --- | --- |
| FDA will implement Capability Maturity Model (CMM) in CBER, CDER, ORA, and OC for PDUFA IT infrastructure and investments, and include other industry best practices to ensure that PDUFA III IT products and projects are of high quality and produced with optimal efficiency and cost effectiveness. This includes the development of project plans and schedules, goals, estimates of required resources, issues, and risks/mitigation plans for each PDUFA III IT initiative. | --- | --- | --- | --- | X |
| Where common business needs exist, CBER, CDER, ORA, and OC will use the same software applications, such as e-CTD software, and COTS solutions. | --- | --- | --- | --- | X |
| Within six months of authorization, a PDUFA III IT five-year plan will be developed. Progress will be measured against the milestones described in the plan. | X | --- | --- | --- | --- |
| -- | Not applicable |
| X | Action due |
A. The term "review and act on" is understood to mean the issuance of a complete action letter after the complete review of a filed complete application. The action letter, if it is not an approval, will set forth in detail the specific deficiencies and, where appropriate, the actions necessary to place the application in condition for approval.
B. Under PDUFA I and II, receipt of a major amendment to original NDAs and BLAs in the last 3 months extended the goal date by 3 months. Under PDUFA III, this extension also applies to efficacy supplements and Class 2 resubmitted NDAs, BLAs, and efficacy supplements. Receipt of a major amendment to a manufacturing supplement in the last 2 months extends the goal date by 2 months (PDUFA III submissions only).
C. A resubmitted original application is a complete response to an action letter addressing all identified deficiencies.
D. Class 1 resubmitted applications are applications resubmitted after a complete response letter (or a not approvable or approvable letter) that include the following items only (or combinations of these items):
1. Final printed labeling
2. Draft labeling
3. Safety updates submitted in the same format, including tabulations, as the original safety submission with new data and changes highlighted (except when large amounts of new information, including important new adverse experiences not previously reported with the product, are presented in the resubmission)
4. Stability updates to support provisional or final dating periods
5. Commitments to perform Phase 4 studies, including proposals for such studies
6. Assay validation data
7. Final release testing on the last 1-2 lots used to support approval
8. A minor reanalysis of data previously submitted to the application (determined by the Agency as fitting the Class 1 category)
9. Other minor clarifying information (determined by the Agency as fitting the Class 1 category)
10. Other specific items may be added later as the Agency gains experience with the scheme and will be communicated via guidance documents to industry.
E. Class 2 resubmissions are resubmissions that include any other items, including any item that would require presentation to an advisory committee.
F. A Type A Meeting is a meeting that is necessary for an otherwise stalled drug development program to proceed (a "critical path" meeting).
G. A Type B Meeting is a 1) pre-IND, 2) end of Phase 1 (for Subpart E or Subpart H or similar products) or end of Phase 2/pre-Phase 3, or 3) a pre- NDA/BLA meeting. Each requestor should usually only request 1 each of these Type B meetings for each potential application (NDA/BLA) (or combination of closely related products, i.e., same active ingredient but different dosage forms being developed concurrently).
H. A Type C Meeting is any other type of meeting.
This appendix updates the detailed review histories of the NDAs and BLAs submitted and approved under PDUFA in FY 2005. Approvals are grouped by submission year and priority designation and listed in order of total approval time. Review histories of all other PDUFA submissions approved prior to FY 2005 can be found in the appendices of the earlier PDUFA Performance Reports that are available at http://www.fda.gov.
| ** | Major amendment was received within 3 months of the action due date, which extended the review timeframes by 3 months. | |
| Action Codes: | AE = Approvable AP = Approved NA = Not Approvable RL = Complete Response TA = Tentative Approval* |
|
* Tentative Approval (TA) is an action given to a product that meets all the requirements for approval; however, it may not be legally marketed in the U.S. until the market exclusivity and/or patent term of the listed reference drug product has expired.
| Receipt Cohort (FY) | Established/Proper Name | Applicant | Approval Time (Months) | Review Goal Met | |
|---|---|---|---|---|---|
| Total Time | Resubmissions (if necessary) | ||||
| 2005 | zidovudine; lamivudine; nevirapine | Pharmacare | 0.4 | Y | |
| 2005 | Influenza Virus Vaccine | GlaxoSmithKline Biologicals | 3.2 | Y | |
| 2005 | Nepafenac | Alcon | 5.7 | Y | |
| 2005 | Vaccinia Immune Globulin Intravenous (Human) | Cangene Corporation | 6.0 | Y | |
| 2005 | fluocinolone acetonide | Bausch & Lomb | 6.0 | Y | |
| 2005 | Tipranavir | Boehringer Ingelheim | 6.0 | Y | |
| 2005 | tigecycline | Wyeth | 6.0 | Y | |
| 2005 | mecasermin (rDNA origin) | Tercica | 6.0 | Y | |
| 2005 | sildenafil citrate | Pfizer | 6.0 | Y | |
| 2005 | emtricitabine | Gilead | 6.0 | Y | |
| 2005 | galsulfase | Biomarin | 6.0 | Y | |
| 2004 | erlotinib hydrochloride | OSI | 3.6 | Y | |
| 2004 | pegaptanib sodium | Eyetech | 6.0 | Y | |
| 2004 | iloprost | Cotherix | 6.0 | Y | |
| 2004 | saquinavir mesylate | Roche | 6.0 | Y | |
| 2004 | pentetate calcium trisodium | CIS | 6.0 | Y | |
| 2004 | pentetate zinc trisodium | CIS | 6.0 | Y | |
| 2004 | entecavir (tablet) | Bristol-Myers Squibb | 6.0 | Y | |
| 2004 | entecavir (solution) | Bristol-Myers Squibb | 6.0 | Y | |
| 2004 | natalizumab | Biogen | 6.0 | Y | |
| 2004 | palifermin | Amgen | 6.0 | Y | |
| 2004 | sodium benzoate; sodium phenylacetate | Ucyclyd | 6.3 | N | |
| 2004 | Vaccinia Immune Globulin Intravenous (Human) | DynPort Vaccine Company, LLC | 8.9 | Y** | |
| 2004 | Clofarabine | Genzyme | 9.0 | Y** | |
| 2004 | trypan blue | Dorc International BV | 13.6 | FDA First Action (AE): 6.0 Sponsor | Y |
| 2004 | trypan blue | Dorc International BV | 13.6 | Response: 1.6 FDA Second Action (AP): 6.0 | Y |
| 2004 | pregabalin | Pfizer | 14.0 | FDA First Action (AE): 8.9 Sponsor | Y** |
| 2004 | pregabalin | Pfizer | 14.0 | Response: 3.2 FDA Second Action (AP): 1.9 | Y |
| 2003 | insoluble Prussian blue | Degussa | 14.3 | FDA First Action (AE): 9.0 Sponsor | Y** |
| 2003 | insoluble Prussian blue | Degussa | 14.3 | Response: 2.5 FDA Second Action (TA): 2.8 | Y |
| 2003 | hyaluronidase | Amphastar | 15.6 | FDA First Action (NA): 6.0 Sponsor | Y |
| 2003 | hyaluronidase | Amphastar | 15.6 | Response: 3.6 FDA Second Action (AP): 6.0 | Y |
| 2002 | micafungin sodium | Fujisawa | 34.6 | FDA First Action (AE): 9.0 Sponsor | Y** |
| 2002 | micafungin sodium | Fujisawa | 34.6 | Response: 18.9 FDA Second Action (AP): 6.7 | Y** |
| 2000 | ziconotide | Elan | 60.0 | FDA First Action (AE): 6.0 Sponsor Response: 7.1 FDA | Y |
| 2000 | ziconotide | Elan | 60.0 | Second Action (AE): 5.8 Sponsor Response: 35.1 FDA | Y |
| 2000 | ziconotide | Elan | 60.0 | Third Action (AP): 6.0 | Y |
| Receipt Cohort (FY) | Established/Proper Name | Applicant | Approval Time (Months) | Review Goal Met | |
|---|---|---|---|---|---|
| Total Time | Resubmissions (if necessary) | ||||
| 2005 | quinine sulfate | Mutual | 9.9 | Y | |
| 2005 | loperamide hydrochloride | Banner Pharmacaps | 10.0 | Y | |
| 2005 | metformin hydrochloride; pioglitazone hydrochloride | Takeda | 10.0 | Y | |
| 2004 | Immune Globulin Intravenous (Human), 10% Solution | Baxter Healthcare Corporation | 9.8 | Y | |
| 2004 | Omeprazole | Santarus | 9.8 | Y | |
| 2004 | Azithromycin | Pfizer | 9.9 | Y | |
| 2004 | Levofloxacin | Sicor | 9.9 | Y | |
| 2004 | dexmethylphenidate hydrochloride | Novartis | 9.9 | Y | |
| 2004 | Exenatide | Amylin | 9.9 | Y | |
| 2004 | leuprolide acetate | Atrix | 9.9 | Y | |
| 2004 | bromfenac sodium | ISTA | 9.9 | Y | |
| 2004 | Paricalcitol | Abbott | 9.9 | Y | |
| 2004 | chlorhexidine gluconate; isopropyl alcohol | Nice-Pak | 9.9 | Y | |
| 2004 | Tetanus Toxoid, Reduced Diphtheria Toxoid and Acellular Pertussis Vaccine, Adsorbed | Aventis Pasteur Limited | 9.9 | Y | |
| 2004 | clindamycin phosphate | Connetics | 10.0 | Y | |
| 2004 | ceftriaxone sodium; dextrose | B Braun | 10.0 | Y | |
| 2004 | Metronidazole | Teva | 10.0 | Y | |
| 2004 | Dapsone | QLT USA | 10.0 | Y | |
| 2004 | Metoclopramide | Schwarz | 10.0 | Y | |
| 2004 | Metronidazole | Dow | 10.0 | Y | |
| 2004 | Ramelteon | Takeda | 10.0 | Y | |
| 2004 | Fluocinonide | Medicis | 10.0 | Y | |
| 2004 | ciprofloxacin hydrochloride | Depomed | 10.0 | Y | |
| 2004 | Amlodipine | Ranbaxy | 10.0 | Y | |
| 2004 | Terconazole | Altana | 10.0 | Y | |
| 2004 | histrelin acetate | Valera | 10.0 | Y | |
| 2004 | levalbuterol tartrate | Sepracor | 10.0 | Y | |
| 2004 | Levofloxacin | Ortho-McNeil | 10.0 | Y | |
| 2004 | donepezil hydrochloride (orally disintegrating tablet) | Eisai | 10.0 | Y | |
| 2004 | donepezil hydrochloride (solution) | Eisai | 10.0 | Y | |
| 2004 | fexofenadine hydrochloride; pseudoephedrine hydrochloride | Aventis | 10.0 | Y | |
| 2004 | Menotropins | Ferring | 10.0 | Y | |
| 2004 | paclitaxel protein-bound particles | American BioScience | 10.0 | Y | |
| 2004 | omega-3-acid ethyl esters | Reliant | 10.0 | Y | |
| 2004 | desloratadine; pseudoephedrine sulfate | Schering | 10.0 | Y | |
| 2004 | brimonidine tartrate | Alcon | 10.0 | Y | |
| 2004 | Oxaliplatin | Sanofi-Synthelabo | 10.0 | Y | |
| 2004 | Tetanus Toxoid, Reduced Diphtheria Toxoid and Acellular Pertussis Vaccine, Adsorbed | GlaxoSmithKline Biologicals | 10.0 | Y** | |
| 2004 | alendronate sodium; cholecalciferol | Merck | 10.5 | N | |
| 2004 | risedronate sodium; calcium carbonate | Procter & Gamble | 11.4 | N | |
| 2004 | Fenofibrate | Reliant | 11.9 | N | |
| 2004 | megestrol acetate | Par | 12.2 | Y** | |
| 2004 | Fenofibrate | Abbott | 12.2 | FDA First Action (AE): 10.0 Sponsor | Y |
| 2004 | Fenofibrate | Abbott | 12.2 | Response: 0.4 FDA Second Action (AP): 1.8 | Y |
| 2004 | Measles, Mumps, Rubella and Varicella Virus Vaccine Live | Merck & Co., Inc. | 12.2 | Y** | |
| 2004 | Aripiprazole | Otsuka America | 12.7 | FDA First Action (AE): 10.0 Sponsor | Y |
| 2004 | Aripiprazole | Otsuka America | 12.7 | Response: 1.0 FDA Second Action (AP): 1.7 | Y |
| 2004 | Meningococcal Groups (A, C, Y, and W-135) Polysaccharide Diphtheria Toxoid Conjugate Vaccine | Aventis Pasteur Inc | 12.9 | Y** | |
| 2004 | doxycycline hyclate | Faulding | 13.0 | Y** | |
| 2004 | clindamycin phosphate | KV | 13.0 | Y** | |
| 2004 | alprazolam | Schwarz | 13.1 | FDA First Action (AE): 10.0 Sponsor | Y |
| 2004 | alprazolam | Schwarz | 13.1 | Response: 1.0 FDA Second Action (AP): 2.0 | Y |
| 2004 | cyanocobalamin | Nastech | 13.1 | FDA First Action (AE): 10.0 Sponsor | Y |
| 2004 | cyanocobalamin | Nastech | 13.1 | Response: 1.2 FDA Second Action (AP): 2.0 | Y |
| 2004 | metformin hydrochloride | Biovail | 13.2 | FDA First Action (AE): 10.0 Sponsor | Y |
| 2004 | metformin hydrochloride | Biovail | 13.2 | Response: 1.3 FDA Second Action (AP): 1.9 | Y |
| 2004 | tramadol hydrochloride | Biovail | 13.7 | FDA First Action (AE): 10.0 Sponsor | Y |
| 2004 | tramadol hydrochloride | Biovail | 13.7 | Response: 1.9 FDA Second Action (AP): 1.8 | Y |
| 2004 | brimonidine tartrate | Allergan | 14.6 | FDA First Action (AE): 10.0 Sponsor | Y |
| 2004 | brimonidine tartrate | Allergan | 14.6 | Response: 2.9 FDA Second Action (AP): 1.7 | Y |
| 2004 | Zolpidem tartrate | Sanofi-Synthelabo | 14.9 | FDA First Action (AE): 10.0 Sponsor | Y |
| 2004 | Zolpidem tartrate | Sanofi-Synthelabo | 14.9 | Response: 3.0 FDA Second Action (AP): 1.9 | Y |
| 2004 | medroxyprogesterone acetate | Pfizer | 15.2 | FDA First Action (AE): 10.0 Sponsor | Y |
| 2004 | medroxyprogesterone acetate | Pfizer | 15.2 | Response: 3.4 FDA Second Action (AP): 1.8 | Y |
| 2004 | tramadol hydrochloride | Biovail | 20.3 | FDA First Action (AE): 10.0 Sponsor | Y |
| 2004 | tramadol hydrochloride | Biovail | 20.3 | Response: 4.3 FDA Second Action (AP): 6.0 | Y |
| 2003 | lidocaine hydrochloride; epinephrine | Empi | 13.0 | Y** | |
| 2003 | tobramycin; loteprednol etabonate | Bausch & Lomb | 15.3 | FDA First Action (AE): 10.0 Sponsor | Y |
| 2003 | tobramycin; loteprednol etabonate | Bausch & Lomb | 15.3 | Response: 3.3 FDA Second Action (AP): 2.0 | Y |
| 2003 | medroxyprogesterone acetate | Pfizer | 17.5 | FDA First Action (AE): 13.0 Sponsor | Y** |
| 2003 | medroxyprogesterone acetate | Pfizer | 17.5 | Response: 2.5 FDA Second Action (AP): 2.0 | Y |
| 2003 | esomeprazole sodium | AstraZeneca | 18.7 | FDA First Action (AE): 10.0 Sponsor | Y |
| 2003 | esomeprazole sodium | AstraZeneca | 18.7 | Response: 2.7 FDA Second Action (AP): 6.0 | Y |
| 2003 | chlorhexidine gluconate | Sage Prods | 19.7 | FDA First Action (AE): 9.9 Sponsor Response: 3.8 | Y |
| 2003 | chlorhexidine gluconate | Sage Prods | 19.7 | FDA Second Action (AP): 6.0 | Y |
| 2003 | albuterol sulfate | Ivax | 21.0 | FDA First Action (AE): 9.9 Sponsor Response: 5.1 | Y |
| 2003 | albuterol sulfate | Ivax | 21.0 | FDA Second Action (AP): 6.0 | Y |
| 2003 | galantamine hydrobromide | Johnson & Johnson | 22.0 | FDA First Action (NA): 9.9 Sponsor Response: 5.2 | Y |
| 2003 | galantamine hydrobromide | Johnson & Johnson | 22.0 | FDA Second Action (NA): 2.0 Sponsor Response: 3.1 FDA | Y |
| 2003 | galantamine hydrobromide | Johnson & Johnson | 22.0 | FDA Third Action (AP): 1.8 | Y |
| 2003 | eszopiclone | Sepracor | 22.5 | FDA First Action (AE): 12.9 Sponsor Response: 3.6 | Y** |
| 2003 | eszopiclone | Sepracor | 22.5 | FDA Second Action (AP): 6.0 | Y |
| 2003 | solifenacin succinate | Yamanouchi | 23.0 | FDA First Action (AE): 9.9 Sponsor Response: 7.1 | Y |
| 2003 | solifenacin succinate | Yamanouchi | 23.0 | FDA Second Action (AP): 6.0 | Y |
| 2003 | ipratropium bromide | Boehringer Ingelheim | 23.3 | FDA First Action (AE): 10.0 Sponsor Response: 7.3 | Y |
| 2003 | ipratropium bromide | Boehringer Ingelheim | 23.3 | FDA Second Action (AP): 6.0 | Y** |
| 2003 | memantine hydrochloride | Forest | 23.6 | FDA First Action (AE): 9.7 Sponsor Response: 11.9 | Y |
| 2003 | memantine hydrochloride | Forest | 23.6 | FDA Second Action (AP): 2.0 | N |
| 2003 | darifenacin hydrobromide | Novartis | 24.7 | FDA First Action (AE): 10.0 Sponsor Response: 8.7 | Y |
| 2003 | darifenacin hydrobromide | Novartis | 24.7 | FDA Second Action (AP): 6.0 | Y |
| 2003 | tetracaine; lidocaine | Zars | 26.6 | FDA First Action (AE): 10.0 Sponsor Response: 10.6 | Y |
| 2003 | tetracaine; lidocaine | Zars | 26.6 | FDA Second Action (AP): 6.0 | N |
| 2003 | calcitonin-salmon (rDNA origin) | Unigene | 29.2 | FDA First Action (AE): 9.9 Sponsor Response: 8.5 | Y |
| 2003 | calcitonin-salmon (rDNA origin) | Unigene | 29.2 | FDA Second Action (AP): 10.8 | N |
| 2003 | insulin detemir | Novo Nordisk | 30.3 | FDA First Action (AE): 9.9 Sponsor Response: 14.6 | Y |
| 2003 | insulin detemir | Novo Nordisk | 30.3 | FDA Second Action (AP): 5.9 | Y |
| 2002 | olopatadine hydrochloride | Alcon | 28.3 | FDA First Action (AE): 9.4 Sponsor Response: 6.5 | Y |
| 2002 | olopatadine hydrochloride | Alcon | 28.3 | FDA Second Action (AE): 5.8 Sponsor Response: 5.2 | Y |
| 2002 | olopatadine hydrochloride | Alcon | 28.3 | FDA Third Action (AP): 1.4 | Y |
| 2002 | lanthanum carbonate hydrate | Shire | 29.9 | FDA First Action (AE): 10.0 Sponsor Response: 10.9 | Y |
| 2002 | lanthanum carbonate hydrate | Shire | 29.9 | FDA Second Action (AP): 9.0 | Y** |
| 2002 | Synthetic conjugated estrogens, B | Duramed | 33.0 | FDA First Action (NA): 13.0 Sponsor Response: 14.3 | N |
| 2002 | Synthetic conjugated estrogens, B | Duramed | 33.0 | FDA Second Action (AP): 5.7 | Y |
| 2002 | oxycodone hydrochloride; ibuprofen | Forest | 35.2 | FDA First Action (AE): 9.9 Sponsor Response: 19.3 | Y |
| 2002 | oxycodone hydrochloride; ibuprofen | Forest | 35.2 | Second Action (AP): 6.0 | Y |
| 2002 | Zolpidem tartrate | Biovail | 40.8 | FDA First Action (AE): 10.0 Sponsor Response: 1.7 | Y |
| 2002 | Zolpidem tartrate | Biovail | 40.8 | FDA Second Action (AE): 2.0 Sponsor Response: 21.3 | Y |
| 2002 | Zolpidem tartrate | Biovail | 40.8 | FDA Third Action (TA): 5.9 | Y |
| 2002 | drospirenone; estradiol | Berlex | 45.4 | FDA First Action (NA): 10.0 Sponsor Response: 17.1 | Y |
| 2002 | drospirenone; estradiol | Berlex | 45.4 | FDA Second Action (AE): 5.9 Sponsor Response: 6.5 | Y |
| 2002 | drospirenone; estradiol | Berlex | 45.4 | FDA Third Action (AP): 5.9 | Y |
| 2001 | lutropin alfa | Serono | 41.3 | FDA First Action (NA): 10.0 Sponsor Response: 26.9 | Y |
| 2001 | lutropin alfa | Serono | 41.3 | FDA Second Action (AP): 4.4 | Y |
| 2001 | gadobenate dimeglumine (single dose vial) | Bracco | 42.9 | FDA First Action (AE): 12.9 Sponsor Response: 16.7 | Y** |
| 2001 | gadobenate dimeglumine (single dose vial) | Bracco | 42.9 | FDA Second Action (AE): 6.0 Sponsor Response: 3.6 | Y |
| 2001 | gadobenate dimeglumine (single dose vial) | Bracco | 42.9 | FDA Third Action (AP): 3.7 | Y |
| 2001 | gadobenate dimeglumine (glass bottle) | Bracco | 42.9 | FDA First Action (AE): 12.9 Sponsor Response: 16.7 | Y** |
| 2001 | gadobenate dimeglumine (glass bottle) | Bracco | 42.9 | FDA Second Action (AE): 6.0 Sponsor Response: 3.6 | Y |
| 2001 | gadobenate dimeglumine (glass bottle) | Bracco | 42.9 | FDA Third Action (AP): 3.7 | Y |
| 2001 | doxazosin mesylate | Pfizer | 46.0 | FDA First Action (AE): 10.0 Sponsor Response: 21.8 | Y |
| 2001 | doxazosin mesylate | Pfizer | 46.0 | FDA Second Action (AE): 6.0 Sponsor Response: 2.2 | Y |
| 2001 | doxazosin mesylate | Pfizer | 46.0 | FDA Third Action (AP): 6.0 | Y |
| 2001 | fenofibrate | Skye | 46.4 | FDA First Action (AE): 10.0 Sponsor Response: 23.3 | Y |
| 2001 | fenofibrate | Skye | 46.4 | FDA Second Action (AE): 8.4 Sponsor Response: 2.7 | Y** |
| 2001 | fenofibrate | Skye | 46.4 | FDA Third Action (AP): 2.0 | Y |
| 2001 | pramlintide acetate | Amylin | 51.1 | FDA First Action (AE): 10.0 Sponsor Response: 20.2 | Y |
| 2001 | pramlintide acetate | Amylin | 51.1 | FDA Second Action (AE): 6.0 Sponsor Response: 9.1 | Y |
| 2001 | pramlintide acetate | Amylin | 51.1 | FDA Third Action (AP): 5.8 | Y |
| 2000 | follitropin beta | Organon | 61.0 | FDA First Action (AE): 10.0 Sponsor Response: 16.8 | Y |
| 2000 | follitropin beta | Organon | 61.0 | FDA Second Action (AE): 8.9 Sponsor Response: 16.2 | Y** |
| 2000 | follitropin beta | Organon | 61.0 | FDA Third Action (AE): 5.8 Sponsor Response: 1.3 | Y |
| 2000 | follitropin beta | Organon | 61.0 | FDA Fourth Action (AP): 2.0 | Y |
| 2000 | fluticasone propionate | GlaxoSmithKline | 63.6 | FDA First Action (WD): 5.4 Sponsor Response: 45.6 | Y |
| 2000 | fluticasone propionate | GlaxoSmithKline | 63.6 | FDA Second Action (AE): 10.0 Sponsor Response: 0.7 | Y |
| 2000 | fluticasone propionate | GlaxoSmithKline | 63.6 | FDA Third Action (AP): 1.9 | Y |
| 1999 | mometasone furoate | Schering | 75.9 | FDA First Action (AE): 10.0 Sponsor Response: 2.0 | Y |
| 1999 | mometasone furoate | Schering | 75.9 | FDA Second Action (AE): 3.4 Sponsor Response: 2.7 | Y |
| 1999 | mometasone furoate | Schering | 75.9 | FDA Third Action (AE): 6.0 Sponsor Response: 35.4 | Y |
| 1999 | mometasone furoate | Schering | 75.9 | FDA Fourth Action (AE): 6.0 Sponsor Response: 4.5 | Y |
| 1999 | mometasone furoate | Schering | 75.9 | FDA Fifth Action (AP): 6.0 | Y |
| 1996 | hydralazine hydrochloride; isosorbide dinitrate | NitroMed | 107.8 | FDA First Action (NA): 12.0 Sponsor Response: 89.8 | Y |
| 1996 | hydralazine hydrochloride; isosorbide dinitrate | NitroMed | 107.8 | FDA Second Action (AP): 6.0 | Y |
1 The count of FY 2005 submissions assumes that all submissions received in the last two months of FY 2005 are filed. When FDA files a submission, it is deemed "complete" by PDUFA definition. FDA makes a filing decision within 60 days of an original application's receipt. All PDUFA review times are calculated from the original receipt date of the filed application.
2 NMEs are a subset of NDAs.
3 Class 1 resubmissions are applications resubmitted after a complete response letter (or a not approvable or approvable letter) that include specific items or combinations of these items (for list of specific items, see Appendix A, page A-7). Class 2 resubmissions are applications resubmitted that include any other items, including any item that would require presentation to an advisory committee.
4 No performance goals for this fiscal year.
5 Includes those with late actions and those still pending whose goal date has passed and which have not had actions.
6 Calculation based only on actions identified as being met or missed. Actions pending within goal were excluded from the calculation.
7 The First Cycle Filing Review Notification goal applies to original NDAs, BLAs, and efficacy supplements only. It does not apply to NDA labeling supplements that contain clinical data, even though these are counted as efficacy supplements for other PDUFA performance purposes. Therefore, the number of filing review notifications for efficacy supplements is less than the total number of efficacy supplements filed (as shown on page 14 ).
8 Receipt of a major amendment in the last 3 months extends the goal date by 3 months. Under PDUFA II (i.e., through FY 2002), this extension applied to original NDAs and BLAs only. Under PDUFA III, it also applies to efficacy supplements and Class 2 resubmitted NDAs, BLAs, and efficacy supplements.
9 Receipt of a major amendment in the last 2 months extends the goal date by 2 months (PDUFA III submissions only).
 Department of Health and Human Services
Department of Health and Human Services 
Food and Drug Administration
| This report was prepared by FDA's Office of Planning in collaboration with the Center for Biologics Evaluation and Research (CBER) and the Center for Drug Evaluation and Research (CDER). For information on obtaining additional copies contact: Office of Planning (HFP-10) Food and Drug Administration 5600 Fishers Lane Rockville, Maryland 20857 Phone: 301-827-5270 FAX: 301-827-5260 This report is available on the FDA Home Page at http://www.fda.gov. |
![]()