


|
| FDA Home Page | CDRH Home Page | Search | A-Z Index |  |
|
A quarterly
bulletin to assist hospitals, nursing homes, and other device user facilities |
|
The following are two cases that the Food and Drug Administration received through its Medical Device Reporting (MDR) system.
Case 1. A family member was given a standard hypodermic syringe with a luer lock that had a removable tip shield to administer an oral suspension to a 5-month-old infant. Inadvertently, the family member administered the medication to the infant without removing the tip shield. The top ejected and lodged in the infant's airway. Attempts to remove the tip shield were unsuccessful. The infant subsequently died.
Case 2. In this injury report, a single dose of liquid antibiotic was sent home with the grandmother of a patient. The medication was put into an oral medication syringe and a protective cap was put on the end of the syringe. When the grandmother administered the medication, she was unaware that the syringe was fitted with a protective cap. When she activated the syringe, the tip ejected and lodged in the infant's throat.
What went wrong?
In the first case, the healthcare practitioner gave the family member a standard hypodermic syringe with a luer lock that was not intended for administrating oral medications. This type of syringe did not provide complete closure around the tip shield and allowed fluid to be drawn up into the syringe with the tip shield in place. The translucent tip shield was similar to the body of the syringe and appeared to be part of the syringe. When the medication was administered with the tip shield in place, the tip shield ejected and lodged in the baby's airway. In the second case, the proper type of syringe was used but the caregiver was not aware that she should remove the protective cap. In both cases, apparently incomplete instructions were given to the caregiver on the safe use of the syringe for oral administration of medications.
What precautions can you take?
Audrey Morrison, RN, BSc in Nursing, is a nurse-consultant in CDRH's Office of Surveillance and Biometrics.
On November 30, 2001, Olympus America, Inc., of Melville, New York initiated a voluntary recall of their bronchoscopes after a medical facility reported that bacteria growing in video bronchoscopes may have been responsible for a patient infection requiring medical treatment. A bronchoscope is a flexible tube with a small light and camera used to evaluate the airway and lungs to diagnose or rule out respiratory problems such as pneumonia and lung cancer.
Olympus issued a recall notification letter to hospitals and medical facilities about the suspect bronchoscopes on November 30, 2001, requesting they be returned to Olympus for modification.
The biopsy port on the video bronchoscope is not intended to be removed from the bronchoscope. However, the medical facility, which reported one illness among patients treated with the bronchoscopes, discovered that the biopsy ports on the bronchoscopes became loose and could be removed by twisting. Olympus America is repairing the bronchoscopes by applying adhesive to the biopsy port and is replacing the plastic biopsy port with a port made from stainless steel.
On February 27, 2002, Olympus issued a second recall notification letter to facilities that did not respond to their first letter.
The following are the Olympus Bronchoscope Models that have been recalled:
BF-40, BF-P40, BF-IT40, BF-3C40, BF-XP40, BF-XT40, BF-240, BF-P240, BF-IT240,
BF-6C240, BF-160, BF-P160, BF-IT160, BF-3C160, BF-XT160.
FDA continues to work with Olympus to get this problem resolved.
If you recently had a lung or airway examination with a bronchoscope and are experiencing respiratory problems, FDA recommends that you contact your physician. In addition, you should report your problem to FDA's MedWatch Reporting Program by telephone, 1-800-332-1088 or on line at http://www.fda.gov/medwatch.
For additional information the recall contact at Olympus is:
Laura Storms-Tyler,
Director, Regulatory Affairs and Quality Assurance
Olympus America Inc.,
Two Corporate Center Drive,
Melville, NY 11747-3157
(631) 844-5688
To contact the Consumer Staff in FDA's Center for Devices and Radiological Health call 1-888-463-6332. When prompted, press 2, press 1, press 3, press 1. To speak to a Consumer Affairs Specialist (from 8:00 AM to 4:30 PM EST), press 5. To request information after business hours, press 4 and leave a message.

| Editor's note: Although the August 14, 2000 Enforcement Priorities Guidance [1] exempts "open-but-unused" single-use devices (SUDs), the effects of resterilizing SUDs raise serious concerns. |
Abstract
Scientists in the Food and Drug Administration's laboratory conducted a study to determine the effect of repeated ethylene oxide (EO) sterilization on sutures that had been opened-but-not-used. Four types of commonly used synthetic absorbable sutures were subjected to 1 and 2 EO resterilization cycles. Knot tensile strength was determined for new sutures and for sutures that had been subjected to 1 and 2 EO resterilization cycles. As has been found with other types of single-use devices, no general conclusions can be made for absorbable sutures. The strengths of different types of sutures increased, decreased, or stayed the same after repeated sterilization. In addition, the inner packages of some sutures were not intact after resterilizing, possibly exposing the sutures to increased humidity. This humidity can produce degradation leading to loss of strength immediately after exposure, after additional shelf aging, and after clinical use.
Introduction
Many absorbable sutures are provided with two layers of packaging: an outer layer that maintains sterility and an inner layer that provides a moisture barrier. Sutures with opened outer packages and intact inner packages, often referred to as opened-but-unused devices [1], are commonly resterilized for reuse although labeled for single use only. Mechanical properties of the polymers used in many sutures can be affected by sterilization methods.
To investigate the possible effects of resterilizing on these single-use devices (SUD), we examined the effect of repeated ethylene oxide (EO) sterilizations on the knot tensile strength of four types of synthetic absorbable sutures. Ethylene oxide sterilization of the sutures was done by the staff at the Materials Management Center at the National Institutes of Health Clinical Center in Bethesda, Maryland.
Methods
The following table summarizes the four types** of size 1 synthetic absorbable sutures that were examined.
| Name | Structure | Composition |
|---|---|---|
| Dexon II | coated, braided | polyglycolic acid |
| Vicryl | coated, braided | copolymers of glycolide and lactide |
| Polysorb | coated, braided | copolymers of glycolide and lactide |
| PDS | monofilament | polydioxanone |
The sutures were sterilized 1 or 2 times with EO (90:10 sterilant mixture with
90% hydrochlorofluorocarbons and 10% EO 75-21-8) for 130 minutes at 54º
- 56º C and 50% - 65% relative humidity, followed by a 12 hour aeration
period. Before each EO cycle, the outer packages were removed and the inner
packages were placed into new outer packages to simulate handling of opened-but-not
used sutures. All packages were carefully handled to avoid mechanically altering
or damaging them.
Mean knot tensile strength was determined for the new samples sutures and after
1 or 2 EO resterilization cycles. Suture strength and knotting of the test strand
was determined according to United States Pharmacopeial (USP) <881>. [2]
All new samples met the USP limit on average knot pull strength given in the
USP official monograph on absorbable surgical suture. [3]
Results
After resterilization using EO, the out-of-package mean knot tensile strength for different types of synthetic absorbable sutures showed a range of responses: increasing, decreasing, or staying the same for different suture types (Figure 1). However, all tested sutures, regardless of EO exposure, met USP limits on average knot pull strength.
Figure 1. Mean knot tensile strength for four types of synthetic absorbable sutures new and after resterilization by EO once or twice. USP limit is 5.08 kgf for size 1 synthetic absorbable sutures
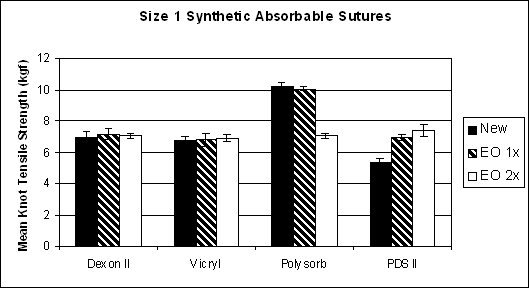
The two-tailed Student's t-test was used for statistical analyses. The test showed no difference in the strength of Dexon II and Vicryl initially and after 1 and 2 EO resterilization cycles. The strength of the Polysorb sutures was not affected after 1 EO cycle but decreased significantly after 2 EO cycles. The PDS II sutures showed a significant increase in strength after 1 EO cycle and an additional increase in strength after 2 EO cycles that was not statistically significant when compared to the 1 EO cycle.
The EO process had readily apparent effects on the suture inner packages.
Figure 2: Resterilized suture inner package with ruptured seal.

Discussion and Conclusions
The testing highlighted a number of concerns related to resterlizing opened-but-unused
sutures.
The most significant conclusion to be drawn from this study is the observation that, as is true for other types of devices [5], it is not possible to make general conclusions about the effects of resterilized on absorbable sutures. Suture strength was not affected for some sutures; others increased, and others decreased in strength with repeated EO sterilization cycles.
References
1. U.S. Food & Drug Administration, Center for Devices & Radiological Health, Office of Compliance. Guidance for Industry and for FDA staff: Enforcement Priorities for Single-Use Devices Reprocessed by Third Parties and Hospitals, August 14, 2000, http://www.fda.gov/cdrh/reuse/1168.html.
2. The United States Pharmacopeial Convention. USP 24-NF 19 Supplement 2. Rockville, MD: The United States Pharmacopeial Convention , July 1, 2000; 2903-4.
3. The United States Pharmacopeial Convention. USP 24-NF 19 Supplement 2. Rockville, MD: The United States Pharmacopeial Convention , July 1, 2000; 2859.
4. Middleton, J.C. and Tipton, A.J., Jr. Synthetic Biodegradable Polymers as Medical Devices. Medical Plastics and Biomaterials. 1998; Vol.5 No. 2. March-April:30-39.
5. Brown, S.A., Merritt, K., Woods, T.O., and Hitchins, V.M., "The Effects of Use and Simulated Reuse on PTCA Balloons and Catheters," Biomedical Instrumentation & Technology 2001;35:312-322.
Terry O. Woods, Ph.D., Stanley A. Brown, D.Eng., Katharine Merritt, Ph.D., Scott G. McNamee, Ph.D., and Victoria M. Hitchins, Ph.D. are scientists in CDRH's Office of Science and Technology.
*Adapted from an article by the authors published in Biomedical Instrumentation & Technology 2001:35:391-94.
**Polysorb, United States Surgical Corporation, Norwalk, CT;
Dexon II, Davis+Geck, Division of American Cyanamid Company, Wayne, NJ; Vicryl,
Ethicon, Inc., Johnson & Johnson, Somerville, NJ; and PDS II, Ethicon, Inc.,
Johnson & Johnson, Somerville, NJ. Representative products and manufacturers
are named for identification only and the list does not imply endorsement by
the U.S. Department of Health and Human Services.
March 15, 2002
To:
Hospital Administrators
Risk Managers
Director, Central Supply
Director, Department of Obstetrics and Gynecology
Director, Department of Surgery
Ambulatory Surgical Centers
I am writing to alert you to a recall of all medical devices manufactured by A & A Medical, Inc. of Alpharetta, GA because they may not have undergone sterilization even though they may be labeled as sterile or ethylene oxide processed. As a result, these devices could cause serious and possibly life-threatening infections.
Other Companies Affected by the Recall
A & A Medical also does business as A&A Medical/Rocket USA, and LifeQuest.
The recall includes all products labeled as sterile and shipped since 1999 nationwide
and internationally.
Please note that these products may be sold by firms other than those listed above. FDA is working to identify all distributors of these products. A list of distributors and the products they receive from A & A Medical is being placed on FDA's web site at http://www.fda.gov/cdrh/recalls/recall31402.html.
We will continue to update this list until all distributors have been identified and listed. In addition, these distributors are being asked to contact all customers who received the affected products.
Products Included in the Recall
This recall includes all products manufactured under the name A & A Medical, Rocket USA, or LifeQuest that are labeled as sterile or as ethylene oxide processed. This firm manufactures many types of Ob/Gyn and surgical devices. The recall includes, but is not limited to curettes (flexible and rigid), uterine dilators, fetal blood samplers, and laparoscopy accessories. A list of known products is attached.
Recommendations
Additional Information
Individuals seeking additional information should call the company at 1-800-424-1234
or 770-343-8400 or contact FDA's Center for Devices and Radiological Health,
Rockville, Maryland at 1-800-638-2041. Additional information regarding this
recall can also be found on the FDA's MedWatch web site at http://www.fda.gov/bbs/topics/NEWS/2002/NEW00799.html.
Should you have questions regarding this letter, please contact, Jan Davis,
Office of Surveillance and Biometrics (HFZ-510), 1350 Piccard Drive, Rockville,
Maryland, 20850, by fax at 301-594-2968, or by e-mail at phann@cdrh.fda.gov.
Additionally, a voice mail message may be left at 301-594-0650 and your call
will be returned as soon as possible.
All of the FDA medical device postmarket safety notifications can be found
on the World Wide Web at http://www.fda.gov/cdrh/safety.html.
Postmarket safety notifications can also be obtained through e-mail on the day
they are released by subscribing to our list server. You may subscribe at http://service.govdelivery.com/service/subscribe.html?code=USFDACDRH_10.
Reporting Adverse Events to FDA
The Safe Medical Devices Act of 1990 (SMDA) requires hospitals and other user facilities to report deaths and serious injuries associated with the use of medical devices, including the recalled devices. We request that you follow the procedures established by your facility for such mandatory reporting.
We also encourage you to report medical device malfunctions. You can report
these directly to the device manufacturer. You can also report to MedWatch,
the FDA's voluntary reporting program. You may submit reports to MedWatch one
of four ways: online at http://www.accessdata.fda.gov/scripts/medwatch/;
by telephone at 1-800-FDA-1088; by FAX at 1-800-FDA-0178; or by mail to MedWatch,
Food and Drug Administration, HF-2, 5600 Fishers Lane, Rockville, MD 20857.
Sincerely yours,
/s/
David W. Feigal, Jr., M.D., M.P.H.
Director,
Center for Devices and Radiological Health
List of Known Ob/Gyn and Surgical Devices Recalled by A & A Medical, Inc.
(This is a partial list as known at the time of this alert.)
Curette (flexible and rigid, all sizes)
Collection set tubing
Aspiration sets
Laminaria
IUD removal instruments
Mucus samplers
Biopsy pipettes/Endometrial sampling sets
Uterine sounds
Pratt dilator set
Ovum forceps
Tenaculum forceps
Needle extenders and guide
Fetal bladder drain
Fetal blood sampler
Harvesting Pump and accessories
Loop/ball electrodes
Laparoscopy accessories
FDA's Center for Devices and Radiological Health (CDRH) is testing a new program to educate healthcare professionals about patient safety issues. "FDA Patient Safety News" is a monthly, 15-minute television program airing on healthcare education networks including Health Science Television Network (HSTN), Long Term Care Network (LTCN), VA's Education Network, and GE TiP TV. The programs provide information on how to protect patients and how to use medical products safely.
The programs provide information about new device approvals, product recalls, safety alerts, and tips for improving patient safety. Some of the episodes may require immediate action by you, such as removing a recalled item from stock or following the recommendations of a Safety Alert or a "safety tip."
Visit the FDA Patient Safety News web site at http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfTopic/psn/index.cfm,
to view the most current program. You will also find more information about
the broadcasts plus links for more information on each episode featured in the
programs.
Topics from May 2002 Broadcast
If you have questions about the web site or any of the programs, write to PSNews@cdrh.fda.gov.
Morgan E. Warner, B.A., is a research assistant in the Center's Office of
Surveillance and Biometrics, Medical Product Surveillance Network Team.
If you would like to be notified electronically (via e-mail) when a new issue of the User Facility Reporting Bulletin is released, you can sign-up for our List Service at:
http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfCDRHNew/listman.cfm
| USER FACILITY REPORTING BULLETIN FDA produces the User Facility Reporting Bulletin quarterly to assist hospitals, nursing homes, and other medical device user facilities in complying with their statutory reporting requirements under the Safe Medical Devices Act of 1990, the Medical Device Amendments of 1992, and the Food and Drug Administration Modernization Act of 1997. The Bulletin's contents may be freely reproduced. Comments should be sent to the Editor. Editor: Nancy Lowe Department of Health and Human Services |
Updated June 21, 2002
![]()
CDRH Home Page | CDRH A-Z Index | Contact CDRH | Accessibility | Disclaimer
FDA Home Page | Search FDA Site | FDA A-Z Index | Contact FDA | HHS Home Page
Center for Devices and Radiological Health / CDRH
