|
|
|
Mefloquine Hydrochloride (marketed as LARIAM and generics)
Pneumonitis
A postmarket safety review of mefloquine, an antimalarial agent, identified cases of pneumonitis or eosinophilic pneumonia associated with use of this drug. This review was prompted by the manufacturer's request to revise the Adverse Reactions-Postmarketing section of the label to include pneumonitis of possible allergic etiology. The product labeling has been updated to reflect this new safety information.
Mefloquine hydrochloride was approved by FDA in 1989. It is widely used as an oral treatment for mild-to-moderate malaria caused by mefloquine-susceptible strains of Plasmodium falciparum (both chloroquine-susceptible and resistant strains) or by Plasmodium vivax. This drug is also approved as a prophylactic treatment for P. falciparum and P. vivax malaria infections, including chloroquine-resistant strains of P. falciparum.1
Mefloquine is usually well-tolerated, although it may cause mild nausea, vomiting, dizziness, insomnia, and nightmares. Rare, severe neuropsychiatric reactions may also occur, including depression, anxiety, psychosis, hallucinations, and seizures.1 There have been five reported cases of mefloquine-associated eosinophilic pneumonia or pneumonitis in the medical literature.2-6
From May 1989 (the date of original approval) to January 2008, FDA has received 13 reports (U.S.-3, non-U.S.-10) of pneumonitis associated with mefloquine therapy. Of the 13 case reports, five are reported in the medical literature. This article summarizes FDA's analysis of these 13 AERS cases.
Reported Cases of Pneumonitis
The 13 cases of pneumonitis reported to AERS involved patients ranging in age from 4-68 years (median age of 53 years). Sixty-nine percent of the patients (9/13) were female. Five patients received mefloquine for treatment of malaria. Six patients were given mefloquine as prophylaxis for malaria. In two cases, information on the underlying condition for which mefloquine therapy was begun was unknown. The median time-to-onset from first administration of mefloquine to respiratory symptoms was 2 days (range 1-84 days). All patients in this case series were hospitalized with various respiratory diagnoses, including pneumonitis, diffuse interstitial pneumopathy, and dyspnea/lung infiltration. Radiographic imaging indicated bilateral lung infiltrates in seven patients. In two cases, fluid from bronchoalveolar lavage (BAL) showed elevated eosinophils and neutrophils. In one patient, lung biopsy revealed an autoimmune interstitial alveolitis. A four-year-old female died after developing pneumonitis. This patient developed symptoms (pulmonary fibrosis and interstitial pneumonitis) after several prophylactic doses of mefloquine. No prior medical history was reported for this patient. Seventy-seven percent of patients (10/13) fully recovered when mefloquine was discontinued. Thirty-eight percent of patients (5/13) improved with systemic corticosteroid therapy. One patient was rechallenged with mefloquine and developed severe pneumonitis. In a number of cases, the recognition of the relationship between the pneumonitis and the use of mefloquine was delayed.
Two representative case reports implicating mefloquine in the development of pneumonitis are described in Box 1. These cases were selected based on the close temporal relationship of the adverse event to the taking of drug, the seriousness of the event, and a positive rechallenge with mefloquine.
In the first case, a case also reported in the medical literature,3 pneumonitis developed one day after initiating mefloquine. Respiratory symptoms reappeared upon rechallenge with the drug. Symptoms gradually waned over a three-week period, most likely attributable to the length of the drug's elimination half-life of two to four weeks.1 The second case describes the development of pulmonary fibrosis and interstitial pneumonitis in a 4-year-old female after she received several prophylactic doses of mefloquine. This patient had no prior medical history of pulmonary disease, and in the absence of other infectious processes, mefloquine was implicated as the causative agent.
|
Box 1
Case 1
Three weeks prior to traveling to Kenya, a 60-year-old woman began prophylactic treatment for malaria with mefloquine (250 mg weekly). On the day following the first dose of mefloquine, she developed a high fever and chills. Empiric antibiotic treatment was started. Four days after her symptoms appeared, she was admitted to the hospital with a fever (101 °F), shortness of breath, cyanosis, myalgia, a nonproductive cough, and headaches. A work-up for the etiology of the infection, including tests for tuberculosis and HIV, was negative. Laboratory blood tests showed a leukocytosis [white blood cell count: 19.9 x103/mm3 (normal 4.3-10 x103/mm3) with 71% neutrophils, 18% lymphocytes and no eosinophils], an elevated C-reactive protein [CRP : 194 mg/dl (normal <3mg/dl)] and an elevated lactate dehydrogenase. A chest X-ray showed bilateral interstitial infiltrates. The patient improved slowly without additional treatment and was discharged after a few weeks with a diagnosis of diffuse interstitial pneumonia of unknown etiology. Four months later, the woman self-started mefloquine prophylaxis (250 mg weekly) ahead of another scheduled trip to Kenya. On the day following the first dose, she once again became severely ill with high fever, and respiratory distress (positive rechallenge). The symptoms were so severe that she was admitted to intensive care unit. Laboratory tests showed results similar to those obtained during her previous hospitalization (leukocytosis, a raised CRP, and elevated LDH). Specifically, there was severe hypoxemia (PaO2: 45mm Hg, pCO2: 32 mm Hg, pH: 7.44) as evidenced by an arterial blood gas analysis. High resolution computed tomography (HRCT) indicated diffuse pulmonary infiltration with ground-glass attenuation. Evaluations for an infectious etiology remained negative. The patient responded well clinically and radiologically to treatment with corticosteroids.
Concomitant medications included aspirin (for atherosclerosis), bisoprolol fumarate (for hypertension), and ciprofibratum (for hyperlipidemia). She had no history of smoking, allergies or pulmonary disease. There was no exposure to animals.
Case 2
A 4-year-old female patient died from pulmonary fibrosis and interstitial pneumonitis after prophylactic treatment with mefloquine. In 2006, prior to traveling, she was started on mefloquine 75 mg per week, an age appropriate dose for the prevention of malaria. The patient had previously taken mefloquine (unknown date). Before the trip, the child was tired and had weight loss. During the trip, she experienced rash and fever (102.2 °F) at night, but was afebrile during the day. She was given an antibiotic for a suspected infection, although subsequent tests revealed no evidence of an infection. Tests revealed no evidence of an infection. On her return, she was hospitalized with suspected inflammatory disease, but no specific diagnosis was given. She was continued on the mefloquine and received corticosteroids which led to her improvement. After 45 days, her general state of health worsened. She started to cough and developed interstitial pneumonitis. Mefloquine was discontinued. A chest radiograph showed bilateral infiltration confluent in the lung. Compared to a previous film, the degree of infiltration had progressed. She was intubated and ventilated due to her rapidly progressive lung failure. A pulmonary biopsy showed autoimmune interstitial alveolitis. She was treated with high dose corticosteroids, plasmaphoresis, and immunoglobulins. After five weeks of extracorporeal membrane oxygenation (ECMO) treatment, the patient died suddenly. An autopsy revealed alveolitis and pulmonary fibrosis.
|
These cases, including a positive rechallenge in one individual, suggest an association between pneumonitis and mefloquine use. Serious cases of pulmonary toxicity occurred when mefloquine was used prophylactically, as well as during the course of treating malaria. One-third of the patients improved following treatment with corticosteroids. Most patients fully recovered upon discontinuing the drug. Antibiotics proved to be an ineffective treatment in many cases suggesting an immune-mediated, rather than infectious, etiology.
Mefloquine-induced pneumonitis is an infrequently reported, but serious, adverse event. FDA will continue to monitor AERS for reports of serious pulmonary toxicity in association with mefloquine.
FDA encourages physicians to:
- Be vigilant if travelers taking mefloquine as prophylaxis or for the treatment of malaria present with symptoms of lung disease or pneumonitis
- Be aware of this infrequent, but serious, adverse event when prescribing mefloquine to avoid delay in diagnosis or treatment
- Report cases of serious pulmonary toxicity in patients taking mefloquine to FDA's MedWatch program at http://www.fda.gov/medwatch
References
- Mefloquine (Lariam) product labeling and Medication Guide.

- Katsenos S, Psathakis K, Nikolopoulou MI, Constantopoulos SH. Mefloquine-induced eosinophilic pneumonia. Pharmacotherapy. 2007;27(12):1767-71.
- Soentjens P, Delanote M, Van Gompel A. Mefloquine-induced pneumonitis. J Travel Med. 2006;13(3):172-74.
- Inoue T, Tanaka E, Sakuramoto M et al. Case of drug-induced pneumonia possibly due to mefloquine (anti-malarial drug). Nihon Kokyuki Gakkai Zasshi. 2005;43(2):103-7.
- Drent M. Drug-induced pneumonia associated with hemizygote glucose-6-phosphate-dehydrogenase deficiency. Eur J Haematol. 1998;61(3):218-20.
- Udry E, Bailly F, Dusmet M et al. Pulmonary toxicity with mefloquine. Eur Respir J. 2001;18(5):890-92.
Back to top
Lenalidomide (marketed as REVLIMID)
Serious skin reactions
A postmarket safety review of lenalidomide identified cases of serious skin reactions, including reports of Stevens-Johnson syndrome (SJS), toxic epidermal necrolysis (TEN), and erythema multiforme (EM), associated with its use. Lenalidomide, an analogue of thalidomide, is an immunomodulatory agent with anti-angiogenic and antineoplastic properties.
In December 2005, lenalidomide 5 mg and 10 mg capsules were approved to treat patients with transfusion-dependent anemia due to low or intermediate-1 risk myelodysplastic syndromes (MDS) associated with a deletion 5q cytogenetic abnormality with or without additional cytogenetic abnormalities.1 The risk classification of MDS is part of an international scoring system for evaluating the prognosis of MDS. This scoring system allows physicians to identify candidates for drug therapy.2 In June 2006, lenalidomide 15 mg and 25 mg capsules were approved for use in combination with dexamethasone for the treatment of multiple myeloma in patients who had received at least one prior therapy for their myeloma. Lenalidomide is considered a teratogenic agent and, in order to prevent pregnancy exposures, is only available under a special restricted distribution program (RevAssist®). Currently, lenalidomide's product labeling does not include information regarding SJS or TEN.1
From the date of its original approval in December 2005 through January 23, 2008, FDA received 14 reports (U.S.-13, non-U.S.-1) of serious skin reactions (i.e., SJS, TEN, and EM) associated with lenalidomide therapy. No additional cases were identified from the literature. This article summarizes FDA's analysis of these cases from the AERS database.
|
Box 1
What are Stevens-Johnson syndrome (SJS), toxic epidermal necrolysis (TEN), and erythema multiforme (EM)?
SJS and TEN are two related, and potentially life-threatening, acute skin disorders that may result from drug exposure. SJS and TEN are characterized by varying degrees of blistering with detachment of the epidermis. For SJS, 10-30% of the body surface area is affected. For TEN, greater than 30% of the body surface area is affected.3 The mortality resulting from these reactions is reported to be between 1-3% for SJS and 10-70% for TEN.4 In industrialized countries, the estimated annual incidence of these adverse events is reported to be 1-2 cases/one million people/year.5
The prodromal phase of SJS/TEN may begin with symptoms of fever, malaise, headache, cough, and rhinorrhea. These initial symptoms may last for 1-3 days before the appearance of flat atypical or purpuric macular lesions. These lesions may then progress to blistering, erosions, and epidermal detachment. The mucosal membranes of the mouth, eye, and genital areas are affected in most patients. Respiratory and gastrointestinal tract lesions may also be present.
Skin lesions may be tender and mucosal lesions may be painful. Early lesions show scattered necrotic keratinocytes in the epidermis. Late lesions show confluent "full-thickness" epidermal necrosis and may eventually form subepidermal bullae.6
SJS/TEN are most often caused by drugs (e.g., anti-infective sulfonamides, non-steroidal anti-inflammatory drugs, anticonvulsants, and allopurinol). Vaccinations, exposure to chemicals and fumigants, and infection with mycoplasma pneumonia are also associated with SJS.6 The greatest risk for developing SJS or TEN is during the first two months of drug therapy.7 Drug causality is usually suspected if the time between the initiation of drug therapy and the onset of SJS and TEN is 4-28 days.6
There are no definitive treatments for SJS and TEN. Supportive care and treatment of specific symptoms are critical. SJS- and TEN-associated mortality may be reduced by early identification and immediate discontinuation of the suspect drug.6 Patients often do better if the discontinued drug has a short half-life (< 24 hours). Corticosteroids, intravenous immunoglobulins, plasmapheresis, cyclophosphamide, cyclosporine, and thalidomide† have been used in the treatment of TEN.7
Erythema multiforme (EM), in contrast to SJS and TEN, is most often caused by the herpes simplex virus. It is a recurrent condition characterized by a limited number of typical or raised target lesions. Blisters may also develop with EM. There is limited oral mucosa involvement. The condition has low morbidity and no mortality.4,6
Footnote
† The study assessing the safety and efficacy of thalidomide in treating TEN was terminated due to a higher mortality with thalidomide compared with the placebo group.7 Thalidomide itself is labeled for SJS/TEN and hypersensitivity reactions.
|
Reported Cases of Serious Skin Reactions
The 14 cases in this report are referred to as SJS/TEN, given the clinical information provided was insufficient to differentiate cases of SJS from TEN. Upon review, three of the 14 reports in this analysis, initially coded in AERS as EM, were re-designated as SJS/TEN as they presented with two or more signs of SJS/TEN (e.g., grade 3 blisters, generalized rash with or without eye and mucosal involvement, and erythema with erosion and crusting of the skin) based on the Common Terminology Criteria for Adverse Events v3.0 (CTCAE; see http://ctep.cancer.gov/reporting/ctc.html). All of these cases required medical intervention.
All dermatological events occurred while patients were taking lenalidomide, with a median time-to-onset of 25 days (range: 3 to 112 days; n=12). Ten patients (71%) were female. The median age of these patients was 70.5 years (range: 46-94 years; n=12). The daily dose reported in 14 cases ranged from 5- 25 mg.
These patients presented with a rash to the arms and/or legs, or to the whole body. Some patients presented with large bullous or vesicular eruptions. In addition, some cases noted the development of pruritis, erythema, burning, facial edema, pain, eruptions in the mouth, around the eyes, or over the abdomen, sore throat, difficulty swallowing, and/or fever. One patient who had a history of a drug allergy to thalidomide (i.e., a rash) experienced what was described as a "Stevens-Johnson type rash" after three days of lenalidomide therapy, suggesting the possibility of a cross-sensitivity between these two drugs.
Thirteen patients received lenalidomide for the approved indications of multiple myeloma (10) and myelodysplastic syndrome (3). One patient was treated with lenalidomide for myelofibrosis (an unapproved indication). Some patients also received treatment with systemic corticosteroids.
Six patients who developed SJS/TEN required hospitalization. Nine of the 14 patients improved or recovered, six of whom also received systemic corticosteroid treatment. There were no rechallenge cases.
There were three deaths reported. Of these cases, one patient died 12 days following hospitalization. Although the cause of death was not provided, there was a diagnosis of SJS at the time of death. The second patient with SJS died eight days following hospitalization. The cause of death was cited as progression of multiple myeloma. The third patient developed TEN following the 4th cycle of lenalidomide (each cycle was 21 days). The patient was hospitalized and the rash resolved five days later. Thirty days following hospitalization, the patient died from progression of multiple myeloma.
Eight patients (57%) reported prior or concurrent therapy with medications that have also been associated with SJS/TEN (i.e., fluoxetine, omeprazole, lansoprazole, esomeprazole, nabumetone, moxifloxacin, escitalopram, sertraline, alprazolam, allopurinol, alendronate, simvastatin, oxcabazepine, and lisinopril). These agents were not listed as co-suspect causes of SJS/TEN. For many of these drugs, however, the date of initiation of treatment was unknown.
Two cases suggesting a role for lenalidomide in the development of serious skin reactions are summarized below (see Box 2). These cases were selected based on a temporal relationship between the adverse event and exposure to drug, the seriousness of the event, and, in one case, a potential cross-sensitivity with thalidomide.
The first case presented describes a patient with a confirmed diagnosis of SJS. The time from the initiation of lenalidomide to the onset of SJS/TEN event was 13 days. This time frame is consistent with the time of onset generally observed for drug-induced SJS/TEN (4-28 days).6 In this case, the patient died from a cause(s) not reported. However, the rash and SJS had not resolved at the time of death.
The second case describes a patient with a history of a thalidomide-induced rash. Three days after receiving lenalidomide, this patient developed a maculopapular rash, urticaria and bullous or vesicular eruptions (SJS-like symptoms).This case suggests a potential cross-sensitivity between lenalidomide and thalidomide.
|
Box 2
Case 1
A 59-year-old female received lenalidomide 25 mg daily for 21 days for multiple myeloma stage III cancer with bone metastasis and renal failure. The patient received dialysis two weeks prior to starting lenolidomide. Twelve days after the first dose of lenalidomide, the patient developed a small rash on her chest. This rash lasted for approximately one week before it eventually resolved (no intervention was reported). During the week following cessation of lenalidomide, the rash returned, worse than before, affecting the whole body, including the face. With the reappearance of the rash, the patient was hospitalized. A diagnosis of SJS was confirmed. Twelve days later, the patient died. Her SJS had not resolved by the time of her death. The cause of death was not provided. Concomitant medications included dexamethasone, eszopiclone, escitalopram, loperamide, a multivitamin, transdermal fentanyl, and hydromorphone.
Case 2
A 67-year-old female received lenalidomide 5 mg orally for three days for treatment of myelofibrosis. Her relevant medical history included a drug allergy to thalidomide (rash).‡ After 3 days of lenalidomide treatment, the patient experienced pruritis, burning, localized maculopapular rash with urticaria, and bullous or vesicular eruptions (Stevens-Johnson type rash). A skin biopsy was not performed. The rash was treated with systemic corticosteroids and resolved. Concomitant medications-some of which had begun more than nine months earlier-included allopurinol, atenolol, folic acid, alendronate, furosemide, glipizide, potassium chloride, levothyroxine sodium, calcium plus Vitamin D, simvastatin, spironolactone, epoetin alfa, docusate sodium, hydroxyurea, metformin, prednisone, and lansoprazole. None of these medications were suspected to be the causative agent in the development of SJS/TEN.
Footnote
‡ Rash, SJS, and TEN are included in thalidomide product labeling.8
|
Although some patients in this case series may have received previous or concurrent medications labeled for SJS/TEN (see above), in all cases, the skin reactions manifested while patients were taking lenalidomide. Some of these patients recovered or improved after discontinuation of lenalidomide. Lenalidomide is an analogue of thalidomide, a drug which is known to cause SJS/TEN, strengthening the probable association between lenalidomide and SJS/TEN in one case. In all cases, the events were serious, required hospitalization and/or medical interventions.
This case series suggests that serious dermatologic reactions, including Stevens-Johnson syndrome and toxic epidermal necrolysis, may occur with lenalidomide therapy.
FDA encourages physicians to:
- Be aware of the possibility of rare serious skin reactions when prescribing lenalidomide
- Discontinue lenalidomide treatment if a skin rash occurs and only resume lenalidomide therapy after appropriate clinical evaluation
- Discontinue and not resume lenalidomide treatment if the rash is exfoliative, purpuric, or bullous, or if Stevens-Johnson syndrome or toxic epidermal necrolysis is suspected
Healthcare professionals and patients should be watchful for skin reactions when using lenalidomide and report any suspected cases to FDA's MedWatch program (http://www.fda.gov/medwatch/).
References
- Revlimid (lenalidomide) product labeling.
- Greenberg P, Cox C, LeBeau MM, et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood. 1997;89(6):2079-88.
- Auquier-Dunant A, Mockenhaupt M, Naldi L, et al. Correlations between clinical patterns and causes of erythema multiforme majus, Stevens-Johnson syndrome, and toxic epidermal necrolysis: results of an international prospective study. Arch Dermatol. 2002;138(8):1019-24.
- Letko GN, Papaliodis DN, Papliodis GN, et al. Stevens-Johnson syndrome and toxic epidermal necrolysis: a review of the literature. Ann Allergy Asthma Immunol 2005;94(4):419-36.
- Pereira FA, Mudgil AV, Rosmarin DM.Toxic epidermal necrolysis. J Am Acad Dermatol. 2007;56(2):181- 200.
- Mockenhaupt M, Viboud C, Dunant A, et al. Stevens-Johnson syndrome and toxic epidermal necrolysis: Assessment of medication risks with emphasis on recently marketed drugs. The EuroSCAR-Study. J Invest Dermatol. 2008;128(1):35-44.
- Wolkenstein P, Latarjet J, Roujeau JC, et al. Randomised comparison of thalidomide versus placebo in toxic epidermal necrolysis. Lancet. 1998;352 (9140):1586-89.
- Thalomid (thalidomide) product labeling.
Back to top
Interaction between amiodarone (marketed as CORDARONE and PACERONE) and simvastatin (marketed as ZOCOR and generics) or simvastatin-combination products (marketed as VYTORIN and SIMCOR)
Amiodarone potentiates the risk for simvastatin-associated rhabdomyolysis
FDA continues to receive reports of rhabdomyolysis in patients given amiodarone in combination with higher doses of simvastatin. Amiodarone is an antiarrhythmic drug indicated to treat certain types of recurrent ventricular arrhythmias. Simvastatin is a 3-hydroxy-methylglutaryl-coenzyme A reductase inhibitor (statin) used to lower cholesterol levels. As with all statins, the risk of rhabdomyolysis is dose-related and increased by high plasma levels of statin. Patients who take amiodarone with simvastatin doses greater than 20 mg daily have an increased risk of rhabdomyolysis. The precise mechanism for this drug interaction is unknown, but stems, in part, from amiodarone's inhibition of the cytochrome P450 3A4 (CYP3A4) enzyme, the same enzyme that metabolizes simvastatin (see Illustration 1). This interaction may result in an increase in the levels of simvastatin in the plasma, potentiating the risk of rhabdomyolysis. Labeling for all of the amiodarone (Cordarone and the generic drug Pacerone)1 and simvastatin-containing products [Zocor2, ezetimibe/simvastatin (Vytorin3) and niacin/simvastatin (Simcor4)] describe this potential risk.
Rhabdomyolysis, a severe form of myopathy, involves injury to and breakdown of skeletal muscles, which in some cases leads to renal failure and death.5 There are multiple etiologies for rhabdomyolysis, including, but not limited to, exposure to certain drugs, including statins.6,7 Healthcare professionals should be aware of the increased risk of rhabdomyolysis when amiodarone is taken concomitantly with doses of simvastatin that exceed 20 mg daily. Prescribers should avoid doses of simvastatin greater than 20 mg per day in patients taking amiodarone (the maximum recommended simvastatin dose is 80 mg daily).
|
Illustration 1
Amiodarone-Simvastatin Interaction - Postulated Mechanism
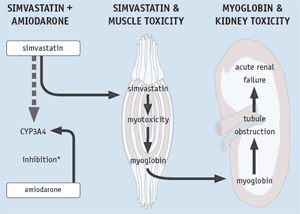 Illustration 1. This illustration depicts a postulated mechanism for the amiodarone-simvastatin interaction, including the subsequent impact of this interaction on skeletal muscle and the kidney. In the first column, amiodarone inhibits the enzyme CYP3A4, limiting simvastatin metabolism (depicted by dashed arrow). By limiting the metabolism of simvastatin, there is an increase in levels of circulating simvastatin in the blood. In the second column, high circulating simvastatin levels may result in myotoxicity in the skeletal muscles (rhabdomyolysis). The rapid breakdown of muscle protein produces excessive levels of myoglobin in the blood. In the third column, myoglobin, now at high circulating levels, reaches the kidneys where it can obstruct renal tubules and lead to acute renal failure. *Amiodarone's direct inhibition of CYP3A4 has been characterized as weak, suggesting that other factors may also contribute to how these two drugs interact.
|
Both the simvastatin and amiodarone labels were changed in 2002 to reflect the increase in risk for myopathy when amiodarone is taken concurrently with simvastatin.1-4 The simvastatin label (Warnings, Precautions and Dosage and Administration sections) specifically indicates that the dose of simvastatin should not exceed 20 mg daily in patients concomitantly receiving amiodarone, and that the combined use of simvastatin and amiodarone at simvastatin doses higher than 20 mg daily should be avoided unless the clinical benefit is likely to outweigh the increased risk of myopathy. The amiodarone label (Precaution section) notes that there is an increased risk for myopathy/rhabdomyolysis when amiodarone is taken in combination with HMG-CoA reductase inhibitors that are CYP 3A4 substrates, such as simvastatin.
Since this labeling change was made, FDA has received 52 additional U.S. reports of rhabdomyolysis associated with the concurrent use of amiodarone and simvastatin. This article summarizes FDA's analysis of these 52 cases from FDA's Adverse Event Reporting System (AERS) database dating from January 1, 2003 to January 1, 2008.
Reported Cases of Rhabdomyolysis
The 52 cases of rhabdomyolysis reported to AERS involved patients ranging in age from 50 to 88 years (median age was 73). Thirty-seven patients (71%) were male and 10 were female (19%). The sex was not reported for the remaining five patients (10%). In half of the reported rhabdomyolysis cases (26/52), amiodarone was being taken in combination with 80 mg simvastatin. Thirteen patients (25%) were taking amiodarone in combination with 40 mg simvastatin, while four patients (8%) were taking amiodarone with 20 mg simvastatin. One patient (2%) developed rhabdomyolysis when taking amiodarone with 5 mg simvastatin. Eight patients (15%) were taking an unknown dose of simvastatin in combination with amiodarone.
Regarding other concomitant medications, 37 patients (71%) were taking medications in addition to amiodarone and simvastatin. These drugs included diuretics (20), beta-blockers (18), angiotensin-converting enzyme inhibitors (16) and insulin (11). Among the concurrent medications taken by these patients, all except for niacin and levofloxacin are either substrates for and/or inhibitors of CYP3A4. These medications included gemfibrozil (9), angiotensin II receptor blockers (3), clarithromycin or levofloxacin (2), protease inhibitors (2), niacin (2), fenofibrate (1), atorvastatin (1), and risperidone (1). The labels of several of these products reflect the risk of rhabdomyolysis when they are used as monotherapy or when administered concurrently with simvastatin.
The mean time interval between the initiation of amiodarone therapy in conjunction with simvastatin (or simvastatin therapy in conjunction with amiodarone) and the onset of rhabdomyolysis was five months (median-2 months). Specifically, 42% of the cases (22) indicated that symptoms of rhabdomyolysis emerged within 2 months of the initiation of concurrent amiodarone-simvastatin therapy. Forty percent of the cases (21) did not report the time interval between the onset of rhabdomyolysis and the initiation of amiodarone-simvastatin therapy.
Ninety-two percent of rhabdomyolysis cases (48) required hospitalization. Twenty-eight percent of the reported cases (15) were considered life-threatening. Ten percent of patients (5) who developed rhabdomyolysis were noted to have become disabled. One death was reported (2%).
Three representative case reports of amiodarone-simvastatin associated rhabdomyolysis are described in Box 1. These cases were selected based on their representation of the demographics and circumstances usually reported with amiodarone/simvastatin-associated rhabdomyolysis. In addition to being reported to AERS, Case 3 has also been published in the scientific literature.8
|
Box 1
Case 1
A 74-year-old male was hospitalized with ventricular tachycardia. While hospitalized, the patient underwent coronary artery bypass graft surgery and was subsequently started on ezetimibe/simvastatin (10/40 mg daily). At the time of discharge, the patient was also prescribed amiodarone 200 mg (to be taken twice daily), aspirin, ramipril, and metoprolol. Three weeks following his discharge from the hospital, the patient complained of extreme muscle weakness. His creatinine level was "highly elevated". A diagnosis of rhabdomyolysis was made. Ezetimibe/simvastastin was discontinued and the patient recovered.
Case 2
A 50-year-old male was hospitalized for coronary artery bypass graft surgery. During his hospitalization, the patient developed atrial fibrillation and was started on amiodarone 400 mg (taken three times daily). The next day, the patient was also started on ezetimibe/simvastatin 10/80 mg daily. Six days following the initiation of simvastatin, the patient experienced progressive leg weakness with a creatine kinase (CK) of 117,400 units/L (for males, normal reference range: 60 to 400 units/L) and a serum creatinine (SCr) of 3.5 mg/dL (normal reference range: <1.5 mg/dL).9 The patient was transferred to the intensive care unit and ezetimibe/simvastatin was discontinued. Three days after the discontinuation of simvastatin, the patient's CK and SCr levels had decreased to 26,700 units/L and 2 mg/dL, respectively.
Case 3
In 2004, a 72-year-old white male was hospitalized complaining of aches and weakness in his thighs. He also noted that his urine was dark for the week prior to his admission. He had a history of diabetes mellitus, hyperlipidemia, hypertension, azotemia, and coronary artery disease. In the summer of 2004, the patient had bypass surgery. Immediately following his bypass surgery, the patient was started on 200 mg amiodarone (taken once daily). One month later, simvastatin (80 mg/day) was prescribed. Other concomitant medications included metformin, enalapril, glimepiride, hydrochlorothiazide and aspirin.
Laboratory testing at the time of the most recent hospital admission indicated a CK level of 19,620 units/L (for males, reference range: 60 to 400 units/L)9 and a SCr of 2.6 mg/dL (normal reference range: <1.5 mg/dL).9 Rhabdomyolysis was suspected and simvastatin was immediately discontinued. Amiodarone was also discontinued four days after discontinuation of simvastatin. Within one day of stopping simvastatin, CK and SCr levels began to fall. Thirteen days after admission to the hospital, CK was 323 units/L and SCr was 1.7 mg/dL.
|
The concomitant use of amiodarone with simvastatin reduces the dose threshold for simvastatin-associated rhabdomyolysis. The cessation of symptoms (and lowering of laboratory values indicative of rhabdomyolysis) after discontinuation of one or both of these drugs indicates that muscle breakdown can be halted and reversed if identified early. Healthcare professionals should be aware that amiodarone may potentiate the risk for simvastatin-associated rhabdomyolysis. Simvastatin doses greater than 20 mg day daily should be avoided in patients taking or initiating amiodarone therapy. Prescribers should consider use of another statin for patients on amiodarone or initiating amiodarone therapy who require simvastatin doses greater than 20 mg daily to meet their lipid goals.
Relevant Website
Patient information sheet on amiodarone
References
- Amiodarone (Cordarone) product labeling.
- Simvastatin (Zocor) product labeling.
- Ezetimibe/Simvastatin (Vytorin) product labeling.
- Niacin extended-release/Simvastatin (Simcor) product labeling.
- Bagley WH, Yang H, Shah KH. Rhabdomyolysis. Intern Emerg Med. 2007;2:210-18.
- Thompson PD, Clarkson P, Karas RH. Statin-associated myopathy. JAMA. 2003;289(13):1681-90.
- The SEARCH Collaborative Group. SLCO1B1 Variants and Statin-Induced Myopathy -- A Genomewide Study. N Engl J Med. 2008 Jul 23. [Epub ahead of print]
- Ricaurte B, Guirguis A, Taylor HC, Zabriskie D. Simvastatin-amiodarone interaction resulting in rhabdomyolysis, azotemia, and possible hepatotoxicity. Ann Pharmacother. 2006;40(4):753-57.
- Kratz A, Ferraro M, Sluss PM, Lewandrowski KB. Case records of the Massachusetts General Hospital. Weekly clinicopathological exercises. Laboratory reference values. N Engl J Med. 2004; 351(15):1548-63.
Back to top
Icodextrin (marketed as EXTRANEAL) and point-of-care glucose monitoring
A Dangerous Drug-Device Interaction
FDA continues to receive reports of adverse events, including fatalities, related to a drug-device interaction associated with the use of icodextrin (Extraneal), a peritoneal dialysis solution, and certain point-of-care glucose monitoring devices that do not use a glucose-specific test strip. Icodextrin is broken down into maltose in vivo. Some test strips used with portable glucose meters cannot differentiate between maltose, glucose and other sugars as they use methods that are not glucose-specific. The test strips associated with this drug-device interaction use glucose dehydrogenase pyrroloquinolinequinone (GDH-PQQ) or glucose-dye-oxidoreductase (GDO) as reagents. Examples of meters currently using these types of test strips include the Accu-Chek (manufactured by Roche) and FreeStyle (manufactured by Abbott) models. We urge healthcare providers and patients to refer to test strip package inserts or to consult the glucose monitoring device and test strip manufacturer(s) to confirm the glucose methodology in any system that is to be used for monitoring patients receiving icodextrin.† A list of toll free numbers for glucose monitor and test strip manufacturers is available at the Baxter Renal Clinical Help Line (1-888-RENAL-HELP).
Due to the presence of maltose in the blood of a patient receiving Extraneal therapy, the use of test strips that are not glucose-specific provides falsely elevated glucose readings. Falsely elevated blood glucose readings may lead to inappropriate insulin administration, which has caused hypoglycemia, coma, and death. Additionally, cases of true hypoglycemia can go untreated if masked by falsely elevated glucose readings.
As indicated in the Warning section of Extraneal's label, blood glucose measurement in patients receiving Extraneal must be done with a glucose-specific method (monitor and test strips) to avoid interference by maltose released from Extraneal. Glucose-specific methods (i.e., methods that are not affected by this interaction) include those that use glucose oxidase, glucose hexokinase, glucose dehydrogenase nicotine adenine dinucleotide (GDH-NAD), or flavin adenine dinucleotide glucose dehydrogenase (FAD-GDH) based reagents.
This drug-device interaction was identified prior to approval of icodextrin and it is described in product labeling. Several safety measures, including patient/healthcare professional education, have been undertaken by the manufacturer. Because FDA continues to receive reports of this adverse event, we are highlighting this drug-device interaction in additional FDA communications to the public. For a complete discussion on this drug-device interaction, including detailed case reports, see the recent FDA communiqué in the Institute for Safe Medication Practices' (ISMP) publication Medication Safety Alert.
Footnote
† A comprehensive list of FDA-cleared GDH-PQQ and GDO blood glucose monitoring systems is not provided because any such list may become outdated or inadvertently exclude systems distributed under multiple trade names. Note, some product lines include test strips that use more than one type of enzyme methodology. Further, manufacturers of GDH-PQQ systems currently on the market may subsequently change to non-GDH-PQQ methodology. Thus, patients and healthcare providers should consult the test strip package insert or contact the glucose monitoring device and test strip manufacturer(s) for information on the type of methodology used.
Relevant Links and Related Information
FDA Patient Safety News (Avoiding Glucose Monitoring Errors in Patients Receiving Other Sugars):
http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/psn/transcript.cfm?show=55#2
http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/psn/transcript.cfm?show=48#4
FDA Center for Biologics Evaluation and Research (Fatal Iatrogenic Hypoglycemia: Falsely Elevated Blood Glucose Readings with a Point-of-Care Meter Due to a Maltose-Containing Intravenous Immune Globulin Product)
ISMP Medication Safety Alert (Be aware of false glucose results with point-of-care testing)
Back to top
|



