|
 |

|
|
 |
|||||
|
|||||||||
|
|
|
|
|
CDER Report to the Nation: 2003 Center for Drug Evaluation and
Research
CDER 2003 Report to the Nation: Adobe Acrobat version of this document ContentsDirector's MessageIntroduction2003 HighlightsMission1 Drug Review
2 Drug Safety and Quality
3 International Activities
4 Communications
Director's MessageLast year, we at the Center for Drug Evaluation and Research worked hard to meet the challenge of promoting and protecting the public health. We were pleased to welcome our new colleagues involved in the review of therapeutic biologics. The dedication, creativity and expertise of all our professional staff have enabled us to meet our everyday deadlines and embark on new initiatives under the framework of FDA’s strategic plan. The strategic plan is very familiar to us because it builds on what we have been doing. The plan outlines a very ambitious effort to achieve five broad priority goals. Efficient, science-based risk managementWe have long led the world in applying the principles of risk management-assessing public health risks, analyzing methods for reducing them and taking appropriate action. With the expanding complexity of our medical challenges and the need to reduce the health risks facing the public at the lowest cost to society, efficient risk management is more important than ever. Our approach to efficient risk management requires the use of the most current biomedical, statistical, managerial and economic science. We aim to achieve quicker access to safe and effective new products and reduce public health risks without unnecessary costs by:
Patient and consumer safetyToo many Americans suffer from preventable adverse events related to pharmaceutical products resulting in human suffering and avoidable medical costs. Consequently, we are enhancing our post-market monitoring, communication and regulatory activities. In addition, one of the most promising new ways we can improve the system for reporting safety problems is to have direct and secure access to relevant modern electronic health information. By supplementing the current passive reporting systems and partnering with healthcare providers and other government agencies, we will develop more innovative and effective information on the risks associated with regulated products. We will help speed the implementation of safer systems for medical care through steps such as:
Better informed consumersInformed consumers represent our nation’s greatest public health asset, because the choices Americans make every day can have a great impact on their own health and the health of the nation. We are undertaking major new efforts to ensure consumers have the most up-to-date, truthful information on the benefits and risks of regulated products. In this arena, we meet two complementary objectives:
Our goal is a well-informed public, empowered to make better choices to improve their health. CounterterrorismWe are working harder and more creatively than ever to speed the availability of the next generation of safer, more effective countermeasures to protect Americans from biological, chemical, nuclear and radiological agents of terrorism. We have two complementary-but necessarily separate-roles in this effort:
A strong FDAOur continued ability to carry out our mission of protecting and advancing America’s health rests squarely on our most important resource: a talented and dedicated staff. More than ever, we rely on a solid cadre of experienced physicians, toxicologists, chemists, statisticians, mathematicians, project managers and other highly qualified and dedicated professionals. The expertise of our professional staff is essential for making our regulatory decisions balanced and fair. A committee of our scientists oversees an extensive program of training, seminars, case study rounds and guest lectures that helps keep our scientists up-to-date on the latest developments in their disciplines and current industry practices. As we look to the challenges ahead, we remain steadfast in our commitment to facilitate the availability of safe and effective drugs, keep unsafe or ineffective drugs off the market, improve the health of Americans and provide clear and easily understandable drug information to health professionals, patients and consumers.
IntroductionWho we areThe Center for Drug Evaluation and Research is America’s consumer watchdog for medicine. We are part of one of the nation’s oldest consumer protection agencies-the Food and Drug Administration. The FDA is an agency of the federal government’s Department of Health and Human Services. We are the largest of FDA’s five centers, with about 1,800 employees. Approximately half of us are physicians or other kinds of scientists. Many of us have experience and education in such fields as computer science, legal affairs and regulatory matters. What we doOur best-known job is to evaluate new drugs for safety and effectiveness before they can be sold. Our evaluation, called a review, makes sure that the drugs we approve meet our tough standards for safety, effectiveness and quality. We also make sure that you and your doctor will have the information you need to use medicines wisely. Once drugs are on the market, we monitor them for problems. Reviewing drugs before marketing. A drug company seeking to sell a drug in the United States must first test it. We monitor clinical research to ensure that people who volunteer for studies are protected and that the quality and integrity of scientific data are maintained. The company then sends us the evidence from these tests to prove the drug is safe and effective for its intended use. We assemble a team of physicians, statisticians, chemists, pharmacologists and other scientists to review the company’s data and proposed use for the drug. If the drug is effective and we are convinced its health benefits outweigh its risks, we approve it for sale. We don’t actually test the drug when we review the company’s data. By setting clear standards for the evidence we need to approve a drug, we help medical researchers bring new drugs to American consumers more rapidly. We also review drugs that you can buy over the counter without a prescription and generic versions of over-the-counter and prescription drugs. Watching for drug problems. Once a drug is approved for sale in the United States, our consumer protection mission continues. We monitor the use of marketed drugs for unexpected health risks. If new, unanticipated risks are detected after approval, we take steps to inform the public and change how a drug is used or even remove a drug from the market. We also monitor manufacturing changes to make sure they won’t adversely affect the safety or efficacy of the medicine. We evaluate reports about suspected problems from manufacturers, health care professionals and consumers. Sometimes, manufacturers run into production problems that might endanger the health of patients who depend on a drug. We try to make sure that an adequate supply of drugs is always available. Monitoring drug information and advertising. Accurate and complete information is vital to the safe use of drugs. Drug companies have historically promoted their products directly to physicians. More and more frequently now, they are advertising directly to consumers. While it is primarily the Federal Trade Commission that regulates advertising of over-the-counter drugs, we regulate unapproved products that may be marketed OTC, including their associated promotional materials, to ensure that they meet applicable approval requirements and are not fraudulent. Advertisements for a drug must contain a truthful summary of information about its effectiveness, side effects and circumstances when its use should be avoided. We are monitoring the industry’s voluntary program to provide consumers useful information about prescription drugs when they pick up their prescriptions. We are watching this program closely to see that it meets its goals for quantity and quality of information. Protecting drug quality. In addition to setting standards for safety and effectiveness testing, we also set standards for drug quality and manufacturing processes. We work closely with manufacturers to see where streamlining can cut red tape without compromising drug quality. As the pharmaceutical industry has become increasingly global, we are involved in international negotiations with other nations to harmonize standards for drug quality and the data needed to approve a new drug. This harmonization will go a long way toward reducing the number of redundant tests manufacturers do and help ensure drug quality for consumers at home and abroad. We also protect drug quality with rigorous manufacturing inspections to ensure compliance with current Good Manufacturing Practice requirements. Why we do itOur present and future mission remains constant: to ensure that drug products available to the public are safe and effective. Our yardstick for success will always be protecting and promoting the health of Americans. Getting consumer input. Protecting consumers means listening to them. We consult with the American public when making difficult decisions about the drugs that they use. We hold public meetings about once a week to get expert, patient and consumer input into our decisions. We also announce most of our policy and technical proposals in advance. This gives members of the public, academic experts, industry, trade associations, consumer groups and professional societies the opportunity to comment before we make a final decision. In addition, we take part in FDA-sponsored public meetings with consumer and patient groups, professional societies and pharmaceutical trade associations. These help us obtain enhanced public input into our planning and priority-setting practices. What is a drug?We regulate drugs used to treat, prevent or diagnose illnesses. However, drugs include more than just medicines. For example, fluoride toothpaste, antiperspirants, dandruff shampoos and sunscreens are all considered "drugs." You can buy some drugs in a store without a prescription, while others require a doctor's prescription. Some are available in less-expensive generic versions. Prescription drugsPrescription medicines must be administered under a doctor’s supervision or require a doctor’s authorization for purchase. There are several reasons for requiring a medicine be sold by prescription:
Over-the-counter drugsYou can buy OTC drugs without a doctor’s prescription. You can successfully diagnose many common ailments and treat them yourself with readily available OTC products. These range from acne products to cold medications. As with prescription drugs, we closely regulate OTC drugs to ensure that they are safe, effective and properly labeled. Generic drugsA generic drug is a chemical copy of a brand-name drug. There are generic versions of both prescription and over-the-counter drugs. Generic drugs approved by the FDA have the same therapeutic effects as their brand-name counterparts. Scientific researchWe conduct and collaborate on focused laboratory research and testing. This maintains and strengthens the scientific base of our regulatory policy-making and decision-making. We focus on:
MissionThe Center for Drug Evaluation and Research promotes and protects public health by assuring that safe and effective drugs are available to Americans. The Food and Drug Administration Modernization Act of 1997 affirmed the center's public health protection role, clarified the FDA's mission and called for the FDA to:
2003 HighlightsWe are pleased to present our eighth performance report. Our work last year offered many Americans new or improved choices for protecting and maintaining their health or new ways to use existing products more safely. Drug reviewChildren and adults with cancer, heart disease and other serious conditions have benefited from our approvals in 2003. We saw improved results from the previous year on overall approvals and decreases in the time it took us to review and approve most applications. A highlight was the approval of 21 new molecular entities with active ingredients never before marketed in the United States. This number increased from the 2002 total of 17. Priority approvals for products of special medical importance increased from 2002 as well. There were 14 priority NDAs and nine priority NMEs, compared to 11 and seven in 2002, respectively.. We met all of our obligations to Congress for prompt and thorough review of drug applications supported by user fees. We increased choices for self-care by approving three medicines for over-the-counter marketing. This included the first switch of a previously prescription-only treatment for frequent heartburn. Our reviews of generic drugs have been prompt and predictable. We approved 263 generic equivalents for prescription or over-the-counter drugs. These included first-time generic approvals of treatments for depression, seizures and high blood pressure. Drug safety and quality All medicines have risks. With modern, state-of-the-art tools and techniques, we are able to detect rare and unexpected risks rapidly and take corrective action quickly. We processed and evaluated more than 370,000 adverse drug events and 3,000 reports of medication errors. We proposed a regulation that called for over-the-counter medicines commonly used in hospitals and all prescription medicines to have a bar code. The rule became final in 2004. We continued to focus on managing risks of marketed medicines, including having a public workshop to gather consumer and scientific input on our proposals for risk management strategies during drug development and after a drug is marketed. We held several public meetings to discuss our effort to promote modernization of pharmaceutical manufacturing. CommunicationsWe met almost weekly with outside experts on difficult scientific and public health issues. Each month, our Internet information site averaged 750,000 visitors and 13.5 million hits. We developed public education campaigns in areas such as antibiotic resistance and buying drugs from outside the United States. Our education program on generic drug quality, specially funded by Congress, has been enormously successful, with many organizations reproducing our materials at no cost to the government. International activitiesWe continued our close work with our colleagues in Japan and the European Union on finding ways to make the drug development process more efficient and uniform. We entered new information-sharing agreements with Canada, Switzerland and the European Union. Quality systemsWe are starting down a long road toward making major improvements to our quality systems to improve the way we do our work. We already have some quality systems and subsystems in place, so we will build on those and add new ones. The basic concepts underlying quality systems are quite simple:
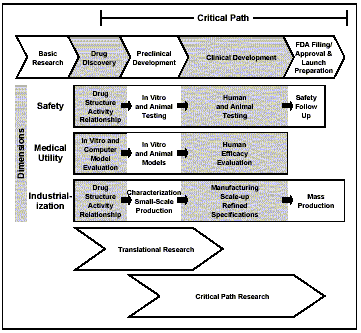
Critical Path InitiativeFDA’s March 2004 report, Innovation or Stagnation?-Challenge and Opportunity on the Critical Path to New Medical Products, provides our analysis of the “pipeline problem.” There is a slowdown-instead of an expected acceleration-in innovative medical therapies reaching patients. The medical product development path is becoming increasingly challenging, inefficient and costly. As a consequence, our mission to ensure the availability of safe and effective medical treatments for Americans that take advantage of the latest science is becoming compromised. In our view, the applied sciences for product development have failed to keep pace with the tremendous advances in the basic sciences. New science is not being used to guide the development process in the same way that it is accelerating the discovery process. To focus the attention of the public, academic researchers, funding agencies and industry, our report identifies:
Critical pathAn idealized “critical path” encompasses the medical product development process. The critical path begins after basic research provides candidate products for development. These products then face successively more rigorous evaluation steps along the path, including:
A striking feature of this path is the difficulty, at any point, of predicting ultimate success with a novel candidate. Recent biomedical research breakthroughs have not improved our ability to identify successful candidates. Critical path dimensionsFrom the earliest phases of preclinical work to commercialization, developers must manage successfully in these three dimensions:
The traditional tools used to assess product safety-animal toxicology and outcomes from human studies-have changed little over many decades and have largely not benefited from recent gains in scientific knowledge. Better tools are needed to identify products that will prove clinically useful and eliminate impending failures more efficiently and earlier in the development process. The current drug discovery process, based on in-vitro screening techniques and animal models of often poorly understood clinical relevance, is fundamentally unable to identify candidates with a high probability of effectiveness. Reaching a more systemic and dynamic understanding of human disease will require major additional scientific efforts as well as significant advances in bioinformatics. The challenges involved in successful industrialization are complex, though highly underrated in the scientific community. Problems in physical design, characterization, manufacturing scale-up and quality control routinely derail or delay development programs and keep needed treatments from patients. Critical path researchThese different types of research support medical product development:
Research needed to improve development toolsWhile the biomedical research community has widened its efforts to include translational research, in our report, we call for a new focus on critical path research. Together with academia, patient groups, industry and other government agencies, we need to embark on an aggressive, collaborative research effort to create a new generation of performance standards and predictive tools that will provide better answers about the safety and effectiveness of investigational products, faster and with more certainty. We at FDA are uniquely suited to take a major role in this effort because of our experience overseeing medical product development, assessment and manufacturing/marketing; our vast clinical and animal databases; and our close interactions with all the major players in the critical path process. The way forwardThis initiative is not a fundamental departure for us, but rather builds on our proven best practices for developing industry guidance and expediting the availability of promising medical technologies. The next steps in this initiative include a series of workshops and meetings, to start development of a National Critical Path Opportunities list and to identify the key priorities. The full report is available at http://www.fda.gov/oc/initiatives/criticalpath/whitepaper.html. Pharmaceutical cGMP InitiativeOur regulatory and quality control systems for pharmaceutical products, known as current good manufacturing practices, have become a gold standard for the world; however, the last comprehensive revisions to these regulations took place nearly a quarter of a century ago. Pharmaceutical cGMPs for the 21st Century is a multi-year Agency effort begun in 2002 to enhance the regulation of pharmaceutical manufacturing and product quality. We evaluated our internal operations under this initiative last year:
cGMP initiative goalsPublic health protection is strengthened by implementing risk-based approaches that focus both industry and our attention on critical areas for improving product safety and quality:
More information is at http://www.fda.gov/cder/gmp/index.htm. Guidances to encourage manufacturing improvementsWe issued one final and four draft guidances to encourage rapid adoption by industry of modern manufacturing practices. These were:
cGMP collaborations
CounterterrorismThe first therapy for those exposed to a terrorism agent is often a drug. We have been taking an aggressive and proactive approach to our role in helping prepare the nation for terrorism attacks. These steps include:
We continue to facilitate development of new drugs and new uses for already approved drugs that could be used as medical countermeasures. We work with other agencies to implement a shelf-life extension program for stockpiled drugs for military use. We gather information on drugs that might be used in response to an attack, including data on manufacturers, bulk suppliers, inventories and lead times for production. We participate in preparedness and response activities to test and establish appropriate communications procedures for emergency situations. We are collaborating with the Centers for Disease Control and Prevention on plans for obtaining post-event outcome data on the use of medical countermeasures. Counterterrorism notable 2003 achievements
Animal efficacy ruleCertain human drugs and biologics intended to reduce or prevent serious or life-threatening conditions may be approved based on animal evidence of effectiveness when human efficacy studies are not ethical or feasible. The regulation, also known as Subpart I for drugs (21 CFR Part 314) or Subpart H for biologics (21 CFR Part 601) applies when:
Counterterrorism scientific researchSome medical countermeasures are stockpiled in tablet form that may be difficult to swallow for infants, small children and others. Two examples are doxycycline, for post-exposure prophylaxis for anthrax, and potassium iodide, for use in emergencies involving radioactive iodine. We used the “electronic tongue” instrument to extend our studies of the stability and palatability of these drugs when crushed and mixed with different foods or drinks. We compared the results to those of human taste panels. We developed an exposure-response model for pyridostigmine, an anti-nerve gas agent, to extrapolate animal efficacy data to a human dose regimen. Counterterrorism biotechnology researchWe have used congressionally mandated special funding to initiate research in several areas relevant to counterterrorism. Our scientists are studying:
By establishing a core of scientists experienced in several areas of bioterrorism, these projects anticipate high-priority regulatory submissions likely to require rapid science-based evaluation. Collaborative research on pneumonic plagueWe are collaborating with the National Institute of Allergy and Infectious Diseases and the U.S. Army Medical Research Institute of Infectious Diseases to investigate the use of the antibiotic gentamicin and other antimicrobials for the treatment of pneumonic plague. Natural history, pharmacokinetic and toxicology studies were performed to support planned efficacy studies using a non-human primate model of pneumonic plague. Shelf-life extension for drug stockpilesOur laboratories perform shelf-life extension testing for drug products stockpiled by the U.S. military and the Strategic National Stockpile. We published the Guidance for Federal Agencies and State and Local Governments: Potassium Iodide Tablets Shelf Life Extension as a draft, and it became final in March 2004. Counterterrorism Internet resourcesWe provides links to the most current information on:
You can find these links at http://www.fda.gov/cder/drugprepare/default.htm. Scientific ResearchWe advance the scientific basis of regulatory practice by developing, evaluating or applying the best, most appropriate and contemporary scientific methods to regulatory testing paradigms. We provide scientific support for reviewer training, regulatory decision making and the development of regulatory policy. We focus on creating a tighter scientific linkage between non-clinical and clinical studies, enhancing methodology for assuring product quality, building databases for improved drug development and review and providing regulatory support through laboratory testing. Linking nonclinical and clinical studies
Biotechnology researchOur new Office of Biotechnology Products was officially transferred in 2003 from the Center for Biologics Evaluation and Research. The office consists of about 80 scientists who are responsible for evaluating therapeutic biotechnology product submissions as well as carrying out scientific research related to biologics regulatory issues.
Our research enhances the ability of our scientist/regulators to evaluate risks and benefits of biotech products, to advise industry on difficult regulatory problems, such as potency assays, and to develop hands-on expertise in the modern technologies used by sponsors of biotech products. Informatics and computational safety analysis
Clinical pharmacology
Pharmaceutical analysis
Laboratory supportLast year our efforts included:
Date created: May 24, 2004
|