|
CDER 2007 Update
Compliance Oversight
We provide comprehensive regulatory coverage of the production and distribution of drug products. We manage inspection programs designed to minimize consumer exposure to defective drug products. We have three basic strategies to meet this goal:
- Evaluate the findings of inspections that examine the conditions and practices in plants where drugs are manufactured, packed, tested and stored.
- Monitor the quality of finished drug products in distribution, through sampling and analysis.
- Monitor drug products to ensure that they comply with applicable approval and labeling requirements.
We identify, evaluate and analyze inspection findings for trends in deficiencies. We publish guidances to assist drug manufacturers and distributors in gaining a better understanding of our regulations. We communicate the expectations of compliance through outreach programs. We review and evaluate for regulatory action all reports of FDA inspections of foreign drug manufacturing facilities. We determine which manufacturers are acceptable to supply active pharmaceutical ingredients or finished drug products to the U.S. market.
Risk-based surveillance sampling of drugs
We monitor the quality of the nation’s drug supply through surveillance and sampling of foreign and domestic finished dosage forms and bulk shipments of active ingredients.
The drug products surveyed are selected according to a risk-based strategy that targets products with the greatest potential to harm the public health. FDA district offices conduct follow-up inspections to determine the cause of sample failures and to assure corrective action by the firms.
Criteria for risk-based sampling
- Microbial/endotoxin concerns.
- Stability concerns.
- Sterility issues.
- Dissolution issues.
- Impurities/contaminants.
- Product quality history.
- Counterfeit drugs.
- History of violations.
Misbranded drugs, unapproved drugs, and unsubstantiated claims
We often encounter misbranded, unapproved, and fraudulent products that make unsubstantiated claims. Consumers may use these products inappropriately. They may use a fraudulent product for treating a serious disease in place of an approved treatment, or they may delay the use of a proper treatment in favor of a fraudulent remedy. Fraudulent products may also contain toxic compounds or other hazardous substances that have the potential to cause serious illness, injury or even death. For these reasons, products that are unapproved, mislabeled, fraudulent, or make unproven claims may pose a significant health risk.
Protecting consumers from unapproved, misbranded or fraudulent drugs
We protect consumers from unapproved, mislabeled, fraudulent or hazardous products. We locate and identify these products on the Internet and other outlets, and we take steps to prevent their sale and to remove them from the market. These steps include issuing enforcement letters and pursuing enforcement actions, such as seizures of violative products and injunctions against firms and individuals. We also work with other federal agencies to coordinate enforcement action against firms and individuals who violate federal law.
We may also take steps to warn the public about unapproved, misbranded and fraudulent products. These steps include issuing press releases and MedWatch alerts to warn consumers about the potential health risks associated with these products. For example, in 2007:
- We issued a warning letter to a firm that marketed several transdermal vitamin therapy products for the prevention and treatment of diseases such as cancer, heart disease, osteoporosis and diabetes.
- We also continued to take enforcement actions against firms that marketed products promoted as dietary supplements for treating erectile dysfunction and enhancing sexual performance, and containing potentially harmful undeclared prescription drug ingredients. Additionally, we continued our efforts to warn consumers about these illegal products by issuing press releases, consumer alerts, articles in consumer oriented publications, and posting information on the FDA website.
Compounded drugs
We generally defer to state authorities regarding the regulation of traditional pharmacy compounding. Traditional pharmacy compounding involves a pharmacist’s customizing of reasonable quantities of drugs in response to a practitioner’s prescription for an individual patient with medical needs that cannot be met by FDA-approved drugs.
Some pharmacies, however, manufacture and distribute compounded drugs in a way that goes beyond traditional pharmacy practice. These pharmacies may make large quantities of unapproved copies of FDA-approved, commercially available drugs when there is no medical need to do so. They may also make these drugs in advance of receiving valid prescriptions. We hold pharmacies that manufacture drug products under the guise of pharmacy compounding to the same federal legal requirements as drug manufacturers.
Furthermore, some pharmacies compound drugs that are contaminated, dangerously weak or strong. Of special concern are the compounding of sterile and more complex dosage forms, such as extended-release drugs, as mistakes in their preparation often lead to serious defects and patient injury. Our steps to protect the public from these products include issuing enforcement letters, referring complaints to state authorities and providing support when states ask. We also pursue enforcement actions, such as seizure of violative products and injunctions against firms and responsible individuals.
Compounding enforcement in 2007
- We warned a firm to stop manufacturing and distributing thousands of doses of unapproved inhalation drugs under the guise of compounding. In a letter to the firm, we identified a range of serious concerns including inadequate quality control, concerns about potency and compounding copies of FDA-approved drugs.
- We sent a warning letter to a repacker of active pharmaceutical ingredients (APIs) that was distributing the API domperidone for use in pharmacy compounding. FDA is concerned with the public health risks associated with domperidone. Domperidone is not an ingredient in any FDA-approved drug. FDA does not sanction its use in pharmacy compounding and will not exercise its enforcement discretion with respect to compounded drugs containing domperidone.
- We warned a firm to stop manufacturing and distributing thousands of doses of injectable drugs under the guise of compounding. FDA inspection revealed that the firm received over 70 adverse event reports associated with the use of its betamethasone injectable drug. In a letter to the firm, we identified a range of serious concerns including inadequate quality control, concerns about potency, and compounding copies of FDA-approved drugs.
Drug imports
We take steps to assure that drugs imported or offered for import into the United States are approved, when required, and do not pose a safety hazard to consumers.
These steps include:
- Preventing entry of products that have been removed from the U.S. market for safety reasons.
- Implementing controls over the importation of drugs, such as human growth hormone, which may pose unique risks to consumers who obtain them outside the legitimate drug distribution system.
Drugs sold without required applications
We identify drugs that are marketed without an approved application. The marketing of products that lack required FDA approval may present safety risks and threatens the U.S. drug development and approval process, as well as the over-the-counter drug monograph system.
We estimate that there are several thousand illegally marketed drug products in the United States, comprised of several hundred unique molecules. In June 2006, we issued a guidance document, Marketed Unapproved Drugs—Compliance Policy Guide, that clearly articulates our expectation that manufacturers of products requiring FDA approval submit applications to show that their products are safe and effective.
The guidance outlines our enforcement policies aimed at efficiently and rationally bringing all such drugs into the approval process and protecting the public health without imposing undue burdens on consumers or unnecessarily disrupting the market. It also creates incentives for manufacturers of marketed, unapproved drugs to seek approval of their products, a process essential to ensuring that physicians prescribe and patients take drug products that are safe, effective, properly manufactured and accurately labeled.
Unapproved drug actions
In 2007 we took action against the following classes of unapproved drugs:
- Trimethobenzamide suppository drug products, used to treat nausea and vomiting in adults and children but lacking evidence of effectiveness.
- Products containing ergotamine, which are used to treat vascular headaches, including migraines. Most of the companies, in addition to marketing these products without FDA approval, omitted from the drugs’ labeling critical warnings regarding the potential for serious, possibly fatal, interactions with certain other drugs.
- Timed-release drug products containing guaifenesin, which are commonly used to relieve cough symptoms. These products lack assurance, through the FDA approval process, that the product releases its active ingredients safely and effectively over the entire time in which the product is intended to work.
- Prescription drug products containing hydrocodone, a narcotic widely used to treat pain and suppress coughs. The action did not affect other hydrocodone formulations, which have FDA approval.
By comparing agency data with a number of private drug data files, we have improved our ability to accurately identify unapproved drug products in the U.S. market. This enhances our ability to develop a rational strategy for bringing all such drugs into the approval process and protecting the public health.
Regulation of OTC drugs
The formulation of OTC drugs and the information that accompanies them or is displayed with them is critical to their safe use.
Approved drug applications and OTC drug monographs define acceptable formulations and the consumer labeling and promotional statements for drugs sold over-the-counter.
- We monitor the statements that accompany these products along with their formulations to make sure that they comply with the appropriate application or final monograph. We also monitor the formulations, labeling and promotional materials associated with over-the-counter drugs marketed without an approved application or final monograph, including fraudulent drugs, and take enforcement actions against these products where necessary.
- We issued warning letters to two prominent firms marketing OTC topical antimicrobials with unsubstantiated claims not covered by FDA’s OTC drug review and without an approved application. Given the extensive advertising and promotion of one such product for use by school children, we issued a press release to alert consumers to the violative nature of that product.
- Following reports of consumer confusion and/or overdose involving several leading brand name and private-label OTC cough-cold drug products packaged with dosage delivery cups, we found that the markings on the cups were inconsistent with the labeled dosage directions. We engaged in discussions with the three manufacturers of these products and all initiated nationwide recalls to consumers.
Assessing data quality, research risks
When obtaining data about the safety and effectiveness of drugs, sponsors rely on high-quality laboratory studies and human volunteers to take part in clinical studies. Protecting volunteers from research risks is a critical responsibility for us and all involved in clinical trials.
We perform on-site inspections to protect the rights, safety and welfare of volunteers and verify the quality and integrity of data submitted for our review. We inspect domestic and foreign clinical trial study sites; institutional review boards; sponsors, monitors, and contract research organizations; laboratories that obtain data; and sites performing bioequivalence studies in humans and preclinical studies in animals.
Our programs to protect volunteers are challenged by increases in the number of clinical trials; the number of sites participating in each clinical study; the types and complexity of products undergoing testing; and the increased number of trials performed in countries with less experience and limited or no standards for conducting clinical research.
Sponsors and clinical investigators protect volunteers by ensuring that:
- Clinical trials are appropriately designed and conducted according to good clinical practices.
- Research is reviewed and approved by an institutional review board.
- Informed consent is obtained from participants.
- Ongoing clinical trials are actively monitored.
- Special attention is given to protecting at-risk populations, such as children and the mentally impaired.
We require sponsors to disclose financial interests of clinical investigators who conduct studies for them. This helps identify potential sources of bias in the design, conduct, reporting and analysis of clinical studies.
Inspections for data quality, research risks in 2007
We conducted 767 inspections in 2007:
- 369 U.S. clinical investigators
- 104 foreign clinical investigators
- 103 institutional review boards
- 23 sponsors, monitors, or contract research organizations
- 46 non-clinical laboratories
- 122 in-vivo bioequivalence studies
The top five deficiencies found during inspections of clinical investigators in 2007 were:
- Failure to follow the protocol
- Failure to keep adequate and accurate records
- Failure to account for the disposition of study drugs
- Failure to report adverse events
- Problems with the informed consent form
International inspections of clinical research
We conducted 104 inspections of clinical research in 28 countries in 2007.
We participate in international efforts to strengthen protections for human volunteers worldwide and encourage clinical investigators to conduct studies according to the highest ethical principles. This includes our work with the International Conference on Harmonization.
Compliance actions in 2007
The Bioresearch Monitoring Program issued 15 warning letters in 2007: ten to clinical investigators, three to institutional review boards, one to a GLP facility, and one to a bioequivalence testing facility. We also initiated the disqualification process for five clinical investigators due to repeated or deliberate violations of the regulations or the submission of false information to the sponsor or FDA.
Bioresearch Monitoring Council
The Bioresearch Monitoring Council was established to coordinate activities and update the program across the five centers in FDA. The Council is composed of representatives of the five centers, Office of Regulatory Affairs (ORA) and the Office of Chief Council, and is conducted under the auspices of the Critical Path Program. The Division of Scientific Investigations is an active member of the Council. We participate on working groups that are charged with updating the compliance policy guides, guidances for clinical investigators, rewriting regulations for the bioequivalence program and the clinical testing of investigational products not conducted under an investigational new drug application. We also participated in the development of guidance for institutional review boards and a guidance regarding questions about the need and application of required information.
We participate as instructors in training courses for ORA investigators for bioresearch monitoring inspections, both at the introductory level and in advanced courses. The courses provide a review of the relevant regulations for each of the five bioresearch monitoring programs, case study review and evaluation and updates on program policies.
Internet resources
More information on data integrity and patient safety is at http://www.fda.gov/cder/offices/dsi/index.htm.
Drug Registration and Listing System
We maintain a database of human drug products in commercial distribution in the United States and all the domestic and foreign establishments involved in their manufacture, repackaging or re-labeling. The Drug Registration and Listing System provides a searchable database for:
- Manufacturers of a specific product.
- Products manufactured by a specific firm.
- Products with a specific ingredient or dosage form.
- Foreign manufacturers in a specific country.
This information helps us administer many key programs, including:
- Postmarketing surveillance.
- User fee assessments.
- Counterterrorism and emergency response.
- Monitoring of drug shortages and availability.
- Identification of products marketed without an approved application.
- Identification of sites for use in our risk-based inspections for good manufacturing practices and adverse drug events.
- Identification of drugs marketed in violation of the law for use in public alerts and enforcement actions.
- In conjunction with commercial databases, identification of marketed unapproved drugs.
Under the FDAAA, an electronic system will replace the current system of submitting paper forms for registration and listing.
Drug Registration and Listing System statistics for 2007
- 14,500 establishment registration forms processed.
- 43,200 drug product listing forms processed.
- approximately 19,500 inquiries answered.
Manufacturing plant inspections
FDA field offices conduct inspections of domestic and foreign plants that manufacture, test, package and label drugs. Before a drug is approved, FDA investigators must determine if data submitted in the firm’s application are authentic and if the plant is in compliance with good manufacturing practices. After a drug is approved, FDA conducts periodic inspections to make sure a firm can consistently manufacture the product with the required quality. We develop compliance programs to guide the investigators in conducting these inspections, and we identify facilities that are high priority for inspection based on their identified risk potential. We provide guidance to explain our current thinking on certain scientific and technical issues.
Prioritizing sites for inspection
Our 2004 white paper, Risk-Based Method for Prioritizing CGMP Inspections of Pharmaceutical Manufacturing Sites—A Pilot Risk Ranking Model, creates a formal risk ranking of manufacturing plants by using an analytical process to:
- Pose a risk question.
- Identify potential hazards and risks.
- Characterize factors that can be used as variables for quantifying risk.
- Mathematically combine the variables to yield an overall risk score.
This program continues to be refined and improved by better evaluation of the risk factors available to us. For example, we added adverse experience reports data to the model in addition to the many data sources already being used. This allows us to maximize our limited resources by focusing our field force on those sites that most affect product quality and safety.
Good manufacturing practice enforcement
We have acted under our regulatory enforcement program to address products not manufactured under current good manufacturing practice regulations. We provide expert technical support that employs science and risk-based principles in applying these regulations. As a result, many corrections are achieved voluntarily or through administrative means. Some corrections, though, require the involvement of the judiciary system. Notable 2007 actions include:
- An inspection of a large repackaging firm uncovered violations whereby some products were mislabeled and some products could have been contaminated by residues from other products packaged using the same equipment. Although the firm recalled many products, FDA officially warned the firm about the importance of implementing promised corrections due to the large scope of their operations—distributing to 47 states and over a 1.4 million patients—and to provide notice should future regulatory action be necessary.
- A manufacturer of highly-potent drugs was warned to improve their equipment cleaning procedures to prevent cross-contamination, and of the importance of organizing operations and drug storage areas to reduce the chance for mix-ups. The firm was also cautioned about the need to properly design and monitor their sterile product filling room.
- A large manufacturer of transdermal drugs was warned about the need to establish a scientifically-sound product specification and of the importance of monitoring all product specifications from batch-to-batch. The firm recalled batches that were not performing well and that were associated with complaints by consumers and health-care practitioners.
- Despite promised corrections, FDA warned a manufacturer of generic prescription drugs about the importance of properly documenting and investigating laboratory deviations and unusual test results.
Domestic drug plant inspections
In fiscal year 2007, FDA field office inspections included:
- 289 preapproval inspections in support of:
- 145 new drug applications
- 177 generic drug applications
- 1,119 current good manufacturing practice inspections
For these, we approved 15 actions:
- 1 seizure
- 14 warning letters
- 67 medical gas inspections
We reviewed 67 medical gas inspections and approved two warning letters.
Biologics license inspections
Our experts conduct preapproval inspections in support of biologics license applications and supplements to them. In fiscal year 2007, there were:
- 10 domestic inspections
- 2 foreign inspections
In other work to ensure the quality of biologics, we reviewed:
- 90 supplements for facilities that did not require an inspection
- 23 annual reports
We held 43 meetings with industry.
Foreign drug inspections
There were a total of 333 inspections. Of these, 208 were PAI/GMP; 89 were PAI only; 24 were GMP; 6 were therapeutic drug product (GMP); and 6 were for cause inspections in 2007.
Council for Pharmaceutical Quality
FDA formed a Council for Pharmaceutical Quality in 2005. The Council oversees policy development and implementation, including the ongoing implementation of internal quality management systems relating to drug quality regulations.
Through our active participation in this program, we have provided the Pharmaceutical Inspectorate advanced training on risk-based approaches to inspections, modern quality systems and the legal and scientific application of good manufacturing practice regulations to manufacturing operations. We certified the first class of these highly trained investigators and are preparing for the next class.
Outreach on drug manufacturing
We published one final and two draft guidance documents that explain our current thinking on certain scientific and technical areas. Guidance documents that were issued to communicate proactively FDA's cGMP (current Good Manufacturing Process) expectations to the pharmaceutical industry include:
- “Testing of Glycerin for Diethylene Glycol.” This final guidance alerts pharmaceutical manufacturers, pharmacy compounders, repackers and suppliers to the potential public health hazard of glycerin contaminated with diethylene glycol (DEG), a poison. FDA has received and continues to receive (most recently in October 2006) reports about fatal DEG poisoning of consumers who ingested medicinal syrups, such as cough syrup or acetaminophen syrup, that were manufactured with DEG-contaminated glycerin. This guidance provides recommendations that will help pharmaceutical manufacturers, repackers, and other suppliers of glycerin, and pharmacists who engage in drug compounding, avoid the use of glycerin that is contaminated with DEG and prevent incidents of DEG poisoning.
- “The Use of Mechanical Calibration of Dissolution Apparatus 1 and 2 - Good Manufacturing Practice (CGMP).” This draft guidance is intended to aid drug manufacturers (including ancillary testing laboratories) in the use of mechanical calibration as an alternate approach to the use of calibrator tablets in calibrating an apparatus used for dissolution testing. This guidance provides references to information on critical tolerances that should be achieved with mechanical calibration.
- “Q10 Pharmaceutical Quality System.” This draft ICH (International Conference on Harmonization) document establishes a new ICH tripartite guideline describing a model for an effective quality management system for the pharmaceutical industry, referred to as the Pharmaceutical Quality System. ICH Q10 describes one comprehensive approach to an effective pharmaceutical quality system that is based on International Organization for Standards concepts, includes applicable GMP regulations, and complements ICH Q8 “Pharmaceutical Development” and ICH Q9 “Quality Risk Management.” ICH Q10 is a model for a pharmaceutical quality system that can be implemented throughout the different stages of a product lifecycle. The content of ICH Q10 that is additional to current GMP requirements is optional.
External Inquiries Program
We communicate our policies and guidance through our External Inquiries Program. Through this program, we coordinate receipt, assignment and response to a large number of foreign and domestic inquiries. Topics ranging from policy issues to current good manufacturing practice questions are covered. In fiscal year 2007, we provided responses to 667 external inquiries.
Drug quality surveillance systems
Our reporting tools help us rapidly identify significant health hazards and quality problems associated with the manufacturing and packaging of medicines. Problems that may affect a medicine’s safety, purity or potency may occur during manufacturing, processing, packing, labeling, storage or distribution.
We evaluate reports and FDA field inspections to identify specific firms with manufacturing quality problems with the most potential impact on public health. We target these candidates for inspection and further product sampling and laboratory analysis. We recommend appropriate corrective actions based upon our analysis of the findings. We may take enforcement action in some cases.
Drug quality reports
Types of reports
- Drug Quality Reporting System. Through MedWatch, we receive reports from consumers and health-care professionals of observed and suspected product quality defects. Our central reporting system assists us in evaluating and prioritizing these data to identify potential manufacturing quality problems and industry trends.
- Field Alert Reports. Applicant holders are required to promptly notify FDA district offices about possible quality and labeling problems that may represent a safety hazard. Experts in FDA district offices evaluate the reports and conduct further investigations when needed.
- Biological Product Deviation Reports. Licensed manufacturers are required to report any event associated with the manufacturing of a therapeutic biological that may affect its safety, purity or potency.
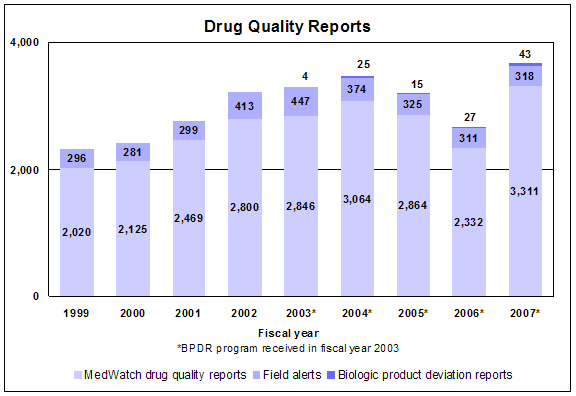
Click image for larger view. Accessible text.
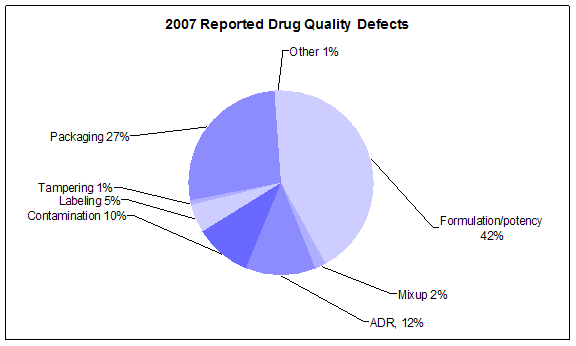
Click image for larger view. Accessible text.
Postmarketing adverse event reporting compliance
We monitor the pharmaceutical industry’s processing of adverse event reports. A firm’s procedures for collection, evaluation and submission of reports may affect the transfer and quality of safety data available for analysis. Our surveillance of industry is based upon the risks associated with specific drug products and specific data processing procedures.
Risk-based inspections
We inspect drug firms’ adverse event reporting based upon risk criteria associated with specific drug products and corporate performance. These include newly marketed drugs, emerging safety signals, previous violations and corporate transitions.
In fiscal year 2007, our field investigators inspected 77 domestic and seven foreign firms to assess compliance with our regulations for adverse event reporting. We sent two firms official notification that they had significant uncorrected deficiencies. We were able to work with 34 firms to obtain voluntary correction of deficiencies identified by our monitoring.
Outreach and education
In addition to our inspectional program for adverse event compliance, we improve safety reporting through educational presentations to industry. Our educational activities include formal presentations at global industry meetings and training for FDA field investigators.
These activities provide industry with a direct opportunity to expand its understanding of reporting requirements and best practices in drug safety, and alert industry to pending regulatory changes. These meetings also serve to expand our own knowledge of industry’s worldwide pharmacovigilance activities.
Next Page
 Back
to Top
Back
to Top  CDER 2007 Update Table of Contents CDER 2007 Update Table of Contents
 PDF requires the free
Adobe Acrobat Reader PDF requires the free
Adobe Acrobat Reader
Date created: July 31, 2008 |





