|
Chapter 24
Identification of Foodborne Bacterial Pathogens by Gene Probes
![]()
DNA Hybridization
The identification of bacteria by DNA probe hybridization methods is based on the presence or absence of particular genes. This is in contrast to most biochemical and immunological tests that are based on the detection of gene products such as antigens or chemical end products of a metabolic pathway.
The physical basis for gene probe tests stems from the structure of DNA molecules themselves. Usually, DNA is composed of two strands of nucleotide polymers wound around each other to form a double helix. These long nucleotide chains are held together by hydrogen bonds between specific pairs of nucleotides. Adenine (A) in one strand binds to thymine (T) in the complementary strand. Similarly, guanine (G) in one strand forms a hydrogen bond with cytosine (C) in the opposite strand. For a discussion of the structure of DNA and nucleic acid hybridization, see Watson et al. (107). An overview (49) of DNA hybridization technology gives a more detailed explanation of hybridization theory, sample preparation, labeling, and formats.
The hydrogen bonds holding the strands together can usually be broken by raising the pH above 12 or the temperature above 95°C. Single-stranded molecules result and the DNA is considered denatured. When the pH or temperature is lowered, the hydrogen bonds are reestablished between the AT and GC pairs, reforming double-stranded DNA. The source of the DNA strands is inconsequential as long as the strands are complementary. If the strands of the double helix are from different sources, the molecules are called hybrids and the process is termed hybridization.
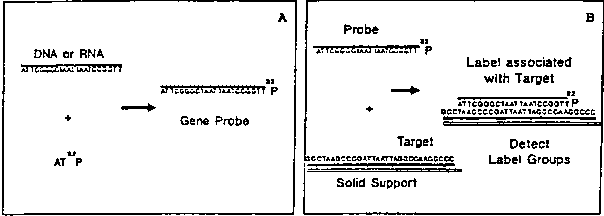
A gene probe is composed of nucleic acid molecules, most often double-stranded DNA. It consists of either an entire gene or a fragment of a gene with a known function. Alternatively, short pieces of single-stranded DNA can be synthesized, based on the nucleotide sequence of the known gene. These are commonly referred to as oligonucleotides. Both natural and synthetic oligonucleotides are used to detect complementary DNA or RNA targets in samples. Double-stranded DNA probes must be denatured before the hybridization reaction; oligonucleotide and RNA probes, which are single-stranded, do not need to be denatured. Target nucleic acids are denatured by high temperature or high pH, and then the labeled gene probe is added. If the target nucleic acid in the sample contains the same nucleotide sequence as that of the gene probe, the probe will form hydrogen bonds with the target. Thus the labeled probe becomes specifically associated with the target (Fig. 1). The unreacted, labeled probe is removed by washing the solid support, and the presence of probe-target complexes is signalled by the bound label.
In addition to DNA, probes and/or their targets can be made of RNA. A number of commercially available gene probe kits use synthetic DNA probes specific for ribosomal RNA targets. DNA:RNA and RNA:RNA hybrids are somewhat more thermally stable than DNA:DNA duplexes, but RNA molecules are quite labile at alkaline pH.
Colony Hybridization
DNA hybridization tests may be performed in many ways. One format, the colony hybridization assay (29,59), will be described here. Generally, an aliquot of a homogenized food is spread-plated on an appropriate agar. After incubation, the colonial pattern is transferred to a solid support (usually a membrane or paper filter) by pressing the support onto the agar surface. Next, the cells are lysed in situ by a combination of high pH and temperature (0.5 M NaOH and/or steam or microwave irradiation), which also denatures and affixes the DNA to the support. The solid support with the attached target DNA is incubated with a 32P- or enzyme-labeled probe. The labeled probe DNA that fails to reform the double helix is removed by washing the probe-target complexes on the support at an appropriate temperature and salt concentration.
Great care must be taken to ensure that the washing temperature is correct; this parameter is usually determined empirically. If the temperature of the washing solution is too high, all the hydrogen bonds between the probe and target may be broken, producing a false-negative result. If the washing temperature is too low, strands of DNA will not match up accurately, and noncomplementary strands may be formed, leading to a false-positive outcome. If the temperature allows only accurately rejoined strands to remain together, the conditions are termed "high stringency." If the temperature is too low, so that mismatched strands exist, the stringency is low. For a review of hybridization using solid supports, see Meinkoth and Wahl (62).
The radioactive probe DNA that is bound to the target on the support is often detected by autoradiography. An X-ray film is placed over the support. Radioactive decays expose the film, so that when it is developed, black spots appear where cells are harboring the same gene as the probe (Fig. 2). If an enzyme-labeled probe is used, a chromogenic substrate is added. Where the probe-associated enzyme is present, a colored spot will develop. Each spot represents a bacterial colony that has arisen from a single cell. The number of cells harboring the target gene in the original sample can be calculated by multiplying the number of spots by the dilution factor.
| Bacteria | Target(a) | Reference |
|---|---|---|
| Campylobacter jejuni | r-RNA | 86 |
| Escherichia coli | Heat-stable toxin (ST) | 74 |
| 103 | ||
| 35 | ||
| Heat-labile toxin (LT) | 104 | 112 | Shiga-like toxins | 41 | Invasive genes | 54 |
| O157:H7 | 20 | |
| Listeria species | r-RNA | 50 |
| L. monocytogenes | Listeriolysin O | 11 |
| Major secreted polypeptide (msp) | 10, 24 | |
| Salmonella species | r-RNA | 7 |
| Shigella species | Invasive genes | 54 |
| Staphylococcus aureus | Enterotoxin B | 78 |
| Vibrio cholerae | Cholera enterotoxin | 57 |
| V. parahaemolyticus | Thermostable direct hemolysin (tdh) | 75 |
| V. vulnificus | Cytotoxin-hemolysin | 112 |
| Yersinia enterocolitica | Cytotoxicity/Sereny | 65 |
| Invasive gene (ail) | 68 | |
| Y. pseudotuberculosis | Invasive gene | 19 |
| a See text under description of individual probes to identify targets. | ||
Target Selection
The first step in developing a gene probe assay is to decide what information is needed. If a particular taxonomic group is to be identified, the probe must be directed toward a gene or region of a gene that is conserved throughout a particular species or genus. On the other hand, one may want to know if a microorganism carrying a particular gene is present. Probes to specific determinants of virulence are useful in assessing a risk to public health posed by bacterial contamination.
Table 1 lists probes that have been used or are of potential use for detecting bacterial pathogens in foods. In the section, "Probes and Their Targets," the development of each probe is described briefly along with what is known about the probe target and its significance. The first probes designed to detect all members of a taxonomic group were constructed by screening randomly cloned DNA fragments. As data on the evolution of ribosomal RNA nucleotide sequences accumulate, probes are being directed toward these targets. Conserved regions can be used to identify large taxons, whereas the variable regions may be unique for a particular genus or species. Furthermore, as a cell contains more than 1000 copies of ribosomal RNA, test sensitivity is increased, because fewer cells are required to produce a positive signal.

Fig. 2. Aliquot of homogenized sample is spread-plated on appropriate medium (cross-hatched area) and incubated until colonies are formed. Colonies are transferred by gentle contact to solid support such as a filter (hatched area). Colony cells are lysed in situ by high pH and/or steam or microwave irradiation, which immobilizes single-stranded target DNA. Filters are then incubated with a labeled gene probe. (In this figure, a radioactive label was used.) Unbound probe is removed by washing the filter at a temperature that allows well-matched double strands to remain joined; poorly matched strands are separated. If DNA from a colony contains the same genetic information as the probe, that area of the filter will become radioactive. Radioactivity is observed as a dark spot on an X-ray film. Count the spots to calculate the number of cells containing specific gene present in the original sample.
Probe Specificity
The relatively short length of synthetic oligonucleotide probes means that they are specific for particular regions of DNA. There is only about 1 chance in 15,000 that a sequence length of 18 bases would appear more than once in the E. coli genome. With a 22-base probe, the chance drops to about 1 in 4 million. To avoid mismatches that reduce specificity, filter washings are conducted at high stringency so that a single base-pair difference between target and probe could not result in hybridization and produce a negative result. Such changes occur as the result of rare mutations. The use of two nonoverlapping probes would significantly reduce the probability of false negatives.
Construction of Probes
Recombinant DNA techniques have made gene probes possible. Probe tests require preparations of relatively pure, specific segments of DNA. The first probes were obtained by inserting these regions into plasmids and transforming the plasmids into the appropriate host cells to increase the amount of probe DNA. Plasmids were purified, and in some cases the inserted fragments were isolated. These cloned, natural DNA probes served quite well, although a considerable amount of effort was required for their production and purification. Through the development of DNA sequencing and automated oligonucleotide synthesis, short (18-30 bases) DNA probes were produced in the laboratory by chemical means. The ready availability of probes considerably expanded their use and application.
Probe Labeling
For probes consisting of cloned DNA fragments, the nick translation method (89) for labeling DNA with radioactivity is very popular. Cloned DNA can also be labeled by a random priming technique (18). Several kits to perform these reactions are commercially available; however, these techniques are unsatisfactory for labeling short oligonucleotides. Oligonucleotide probes are usually labeled on the 5' end with 32P, using bacteriophage T4 polynucleotide kinase and gamma-AT 32P (88). Although radioactive gene probes seem to have the greatest sensitivity in colony hybridization procedures, they are a potential biohazard, and disposal of radioactive waste can be expensive.
Many schemes are being examined for the nonradioactive labeling of gene probes. Some of these techniques have been incorporated into commercial tests designed to signal the presence or absence of a particular gene. For example, alkaline phosphatase has been conjugated to synthetic oligonucleotides without affecting the kinetics or specificity of the hybridization reaction (40).
The Polymerase Chain Reaction
At present it is not practical to use gene probes to detect bacteria directly in foods. Current methods require about 105-106 copies of the target sequence to yield a clear, positive result. To make this number of copies, cells are allowed to replicate in liquid media or to form colonies on agar plates. The growth period is usually overnight, adding 16-24 hours to the length of the test.
It is now possible to amplify specific DNA segments enzymatically to a million-fold in 1-3 hours. This process is called the polymerase chain reaction (PCR) (92). The reaction has been automated by using a thermostable enzyme and a programmable heating block (93). Because of the rapid amplification of target DNA, 1-day probe tests may be developed in the near future. A review of PCR has been published (16). PCR has been used to detect enteroinvasive E. coli and Shigella spp. (54), V. vulnificus (37) Hepatitis A virus (see Chapter 26), and V. cholerae (see Chapter 28) in foods.
Description of Probes and Their Development
The design and construction of gene probes requires careful scientific experimentation and a series of complex decisions. A first step is to determine if the gene probe is to be targeted to a particular pathogenic strain or to an entire taxonomic group. A target must be chosen so that all of the microorganisms to be detected contain such a gene. For probes designed to detect all members of a genus or species, ribosomal RNA has been a popular target because it contains both conserved and variable regions. If a pathogenic strain is sought, a probe is usually targeted to a virulence factor gene responsible for causing disease. A considerable amount of research is needed to identify the genes involved and the role they play in pathogenesis.
Probes and Their Targets
Campylobacter jejuni: Ribosomal RNA
A probe that is specific for C. jejuni ribosomal RNA genes has been developed (86,87) and is available commercially. A pool of randomly selected and tested chromosomal fragments is also specific for C. jejuni, but the target has not been reported (83).
Escherichia coli: Heat-labile enterotoxin genes
The heat-labile enterotoxins (LT) of E. coli are a closely related group of proteins; they are distinguished from heat-stable enterotoxins (ST) by being immunogenic and are inactivated by heating at 60°C for 10 Min (31). The toxins stimulate adenylate cyclase (30) and can be detected by tissue culture assays of Chinese hamster ovary cells (30) or mouse Y-l adrenal cells (13). Using these tests, So et al. (102) localized and cloned the structural gene for LT; Dallas et al. (8) recloned a smaller fragment into plasmid pEWD299. Although there are several different genes for LT, as evidenced by their nucleotide sequences (56,73,103,110,111), they all share a significant amount of genetic similarity. The region of the LT genes chosen as a gene probe target is identical in each of these genes, so that all strains with the genetic potential to produce LTs should be detected.
The LT probe, eltA11, is a 20 base synthetic oligonucleotide that encodes amino acids 45-51 of the A subunit of the E. coli LT (111).
E. coli: Heat-stable enterotoxin genes
The heat-stable enterotoxin (ST) of E. coli is distinguished from LT (above) by heat stability and lack of immunogenicity. It can be detected by the suckling mouse bioassay (12) and acts by stimulating guanylate cyclase (22). There are at least two different types: ST I (also known as STa and STP) and ST II (also known as STb and STH). The latter toxin is not active in the infant mouse assay. These genes have been cloned and the nucleotide sequences of the region encoding STa and STb have been determined (74,82,101).
The STP probe is a 22 base synthetic oligonucleotide for the toxin type strain first isolated from pigs. It targets the region of the gene that encodes amino acids 4-12 of the toxin protein.
The STH probe is targeted to the ST elaborated by a strain of E. coli isolated from a human. The probe is also 22 bases long and targets the region of the STH that encodes amino acids 19-26 of the toxin.
Both of these probes have been tested for their specificity, and data are available on their ability to detect a few ST-producing cells against a high level of ST-negative microorganisms (35). The reliability of the colony hybridization technique with oligonucleotide probes was tested by collaborative study, using pure cultures of strains harboring the STH or STP genes (36).
Enteroinvasive Escherichia coli (EIEC) and Shigella: Invasive gene
Some strains of E. coli invade colonic epithelial cells, multiply intracellularly, and spread intercellularly, causing a dysenteric enteritis similar to that caused by Shigella (15). However, an important difference is that the infectious dose for Shigella may be as low as 1-10 organisms, whereas 108 EIEC cells are necessary to cause disease. A number of genetic determinants that encode virulence factors of EIEC and Shigella spp. are located on a large [220 kilobase (kb) pair] invasion plasmid (96). Loss of this virulence plasmid renders the bacterium avirulent (97). A 17 kb EcoRI fragment was used as a hybridization probe to detect invasive Shigella species and EIEC (4).
Small and Falkow (100) demonstrated that a 2.5 kb pair HindIII fragment of the large plasmid is required for invasion of human epithelial cells. Plasmid DNA involved in the invasion of HeLa cells by S. flexneri has also been cloned (61). These regions of the plasmid have been sequenced and are genetically similar (54). A probe from this region of the plasmid is specific for tissue culture cell-invasive EIEC and Shigella. Such probes also identify strains that are invasive in the guinea pig eye assay (98). Of 41 probe-positive isolates tested by the guinea pig method, 2 were negative, indicating that a few strains may be invasive in tissue culture assays but not in tests that require a greater number of pathogenic determinants (108). A synthetic probe of 18 bases has been constructed. Its target is within a gene that encodes for a virulence factor.
Enterohemorrhagic E. coli (EHEC): Shiga-like toxin (SLT) genes
Human illnesses ranging from simple diarrhea to hemorrhagic colitis and hemorrhagic uremic syndrome have been associated with strains of E. coli that produce moderate to high levels of Shiga-like toxins (SLTs). Strains of E. coli serotype O157:H7 are the most significant pathogens associated with hemorrhagic colitis; strains of serotype O26:H11 are also classified as EHEC. More than 50 other serotypes of E. coli that produce SLTs have been identified, but the correlation of these serotypes with disease is uncertain. Two related but distinct cytotoxins, SLT I and SLT II, have been characterized. Individual strains produce one or both cytotoxins. For example, E. coli O157:H7 produces SLT I, SLT II, or both, whereas O26:H11 produces only SLT I. The DNA sequences of SLT I and SLT II structural genes have been published, and analysis shows that SLT I has 99% homology with the Shiga toxin gene of S. dysenteriae type 1, but SLT II has only 60% homology (41). Two synthetic oligodeoxyribonucleotide probes were prepared from sequence data from the A-subunit regions of the SLT I and SLT II genes (nucleotides 473-490 and 472-490, respectively). HC agar (M62) (see ref. 105) was the selective medium chosen to screen isolates and foods for E. coli strains that carry the SLT gene because the growth of E. coli O157:H7 is less inhibited on HC agar than on other selective media. HC agar contains NaCl and a lower concentration of bile salts No. 3. Its plating efficiency of a strain of E. coli O157:H7 at 37 and 43°C for 17 h was similar to that of plate count agar. Plating efficiencies of other E. coli serotypes that carry the SLT gene have not been determined. The modified enrichment procedure of Doyle and Schoeni (14) is included in the method for detection of low level contamination of foods. For additional information about enterohemorrhagic strains of E. coli, see the review by Karmali (46).
Enterohemorrhagic E. coli (EHEC): O157:H7 serotype-specific probe
The fluorogenic MUG assay for E. coli is based on the activity of the -glucuronidase (GUD) enzyme, which is encoded by the uidA gene in E. coli. Although isolates of serotype O157:H7 are negative with the MUG assay, genetic studies have shown that this EHEC serogroup also contains uidA gene sequences for the GUD enzyme (21). Sequencing analysis has determined that the uidA gene of O157:H7 serotype contains several base mutations; therefore, it is not identical to the uidA gene of MUG assay (+) E. coli. Although the base mutations in the uidA allele of O157:H7 do not appear to be responsible for the absence of the MUG phenotype, one of the base changes was found to be conserved among the O157:H7 serogroup. An oligonucleotide probe, PF-27, directed to this base alteration was developed and determined to be specific only for EHEC isolates of serotype O157:H7. Other SLT-producing EHEC and other pathogenic E coli or enteric bacteria failed to hybridize with PF-27 (20).
Listeria monocytogenes: Invasion-associated protein (iap) and hemolysin (hly) genes
Of the seven Listeria species that have been isolated from a variety of foods, including dairy, vegetable, meat, and poultry products, only L. monocytogenes has been implicated in human disease. Genetic and physiological studies have incriminated an extracellular hemolysin as one of the virulence factors in L. monocytogenes (5,26,47). This hemolysin (also called listeriolysin O or alpha-listeriolysin) has been cloned and sequenced (11,55,64). Several oligonucleotides (including AD13) were constructed by using the sequence of the listeriolysin O gene (64) and can specifically identify L. monocytogenes in foods by colony hybridization (11,70).
A 5.3 kb DNA fragment encoding a 60 kilodalton (Kdal) protein (msp) associated with hemolytic activity has been cloned (24). Kuhn and Goebel (53) reported the cloning and sequencing of a gene (iap) whose product (a 60 Kdal protein) may be involved in the uptake of L. monocytogenes by nonprofessional phagocytes. Sequence analysis revealed that the msp and iap share extensive homology, which indicates that msp and iap may be the same gene (51). An internal region of this gene was sequenced (Datta, unpublished results) and a synthetic probe, AD07, was used to identify and enumerate L. monocytogenes in a number of foods (9,10,34). Thus, either AD07 (for the iap gene) or AD13 (for the hly gene) can be used to detect and enumerate L. monocytogenes in foods. To avoid false-negative results because of "silent" mutations in the gene (nucleotide changes that affect DNA probe binding but do not change the gene function), both probes should be used in combination (designated AD713).
Salmonella species:
Originally, several restriction endonuclease fragments selected randomly from the Salmonella chromosome were used as probes to identify members of the genus (23). Although these molecules served as specific probes, the role played by the target genes was never reported. More recently, probes were developed for regions of the bacterial ribosomal gene that are unique for salmonellae. These probes were used to develop a commercial kit that also uses a nonisotopic labeling and detection system (7).
Staphylococcus aureus: entB probe
Six groups (A, B, C1, C2, D, and E) of related enterotoxins associated with pathogenicity are elaborated by some strains of S. aureus and can cause symptoms of staphylococcal food poisoning if ingested (1). The genes for enterotoxins A, B, C1, and E (entA, entB, entC1, and entE) have been cloned and sequenced (2,3,6,43,85).
Three synthetic oligonucleotide probes were synthesized according to the sequence of the entB gene and used to test 210 strains of S. aureus (78). One probe was specific for entB; the others hybridized with strains producing enterotoxin C. The former probe was used to detect EntB-producing S. aureus in artificially contaminated crabmeat (Trucksess and Williams, manuscript in preparation). Although the nucleotide sequences of enterotoxin genes for groups A, C1, and E are known, synthetic probes have not been reported.
Vibrio cholerae: Cholera toxin
The classical cholera enterotoxin (CT) is a major virulence factor in pathogenic strains of V. cholerae. The mechanism of action and immunological reactivity is quite similar to the LT of E. coli. Genes encoding this multisubunit protein were cloned and sequenced (57,58,63). Non-O1 V. cholerae enterotoxin genes are apparently similar to classical CT (33). Two sequences from the A subunit structural gene for production of the classical enterotoxin are used as probes to detect the CT gene: ctxA11 (bases 702-721) and ctxA12 (bases 718-735).
Vibrio parahaemolyticus: Thermostable direct hemolysin
An important foodborne pathogen often associated with seafood, V. parahaemolyticus can produce a thermostable direct hemolysin (TDH) (95), also referred to as the Kanagawa phenomenon-associated hemolysin (69). This phenotype is commonly associated with strains isolated from humans with gastroenteritis but rarely found in environmental isolates (44). It is not yet known if this hemolysin is a virulence factor, but epidemiological evidence suggests that it is. The gene for the hemolysin has been cloned and sequenced (45,75,106). The specificity of both the cloned probes (76) and a synthetic oligonucleotide probe, tdh3 (77) has been established. The tdh3 probe is 18 bases long and its target encodes amino acids 122-128 of the tdh gene.
Vibrio vulnificus: Cytotoxin/hemolysin
V. vulnificus has been implicated as a cause of human infections and septicemia. The primary source of infection appears to be raw or undercooked seafood, especially raw oysters (71). This lactose-positive vibrio produces a cytotoxin/hemolysin which was implicated as a virulence factor (28), and the gene has been cloned (109). A 3.2 kb DNA fragment carrying the structural gene for this protein is a specific probe for V. vulnificus (72) and has been sequenced (112). One synthetic probe (VV6) exhibited 100% specific for 166 laboratory and environmental strains of V. vulnificus (FDA Contract No. 223-84-2031, Task XIII).
Yersinia pseudotuberculosis: Invasive gene (INV-3)
A chromosomal gene of Y. pseudotuberculosis, inv, which plays an integral part in Yersinia pathogenicity, has been cloned and sequenced (38,39). Oligonucleotide probe INV-3, based on published inv sequence (39) is 21 nucleotide bases long and targeted to a region 200 base pairs away from the 5' terminus of the inv gene of Y. pseudotuberculosis (19). Tests of INV-3 using Southern and colony hybridizations were compared with HeLa cell invasion studies and shown to be specific only for invasive Y. pseudotuberculosis isolates. Although there are homologous sequences between the inv genes of Y. enterocolitica and Y. pseudotuberculosis, this homology is not detectable by INV-3.
Yersinia enterocolitica: Chromosomal invasion gene (PF-13)
The genes responsible for mammalian cell invasion are also carried on the chromosome in Y. enterocolitica (66). Unlike Y. pseudotuberculosis, however, Y. enterocolitica has two loci that encode the invasion phenotype. The inv locus, homologous to the inv gene of Y. pseudotuberculosis, allows high level invasion of several tissue culture cell lines, whereas the ail gene shows more host specificity (66). Analysis of Yersinia serotypes and species using cloned probes from inv and ail showed that all disease-causing isolates are tissue culture-invasive; all these isolates reacted with the AIL gene probe (67). The INV probe reacted with both tissue culture-invasive and noninvasive isolates; however, recent evidence suggests that the inv in these noninvasive strains may not be expressed. The oligonucleotide probe PF13 is targeted specifically to a region 60 base pairs away from the 3' terminus of the ail gene of Y. enterocolitica. The probe is 18 nucleotides in length. A comparison of colony and Southern hybridization studies of 150 yersiniae and non-yersiniae isolates and HeLa cell invasion studies showed that PF13 hybridized only with invasive Y. enterocolitica isolates (19).
Yersinia enterocolitica: Plasmid gene (SP-12)
All pathogenic Yersinia species carry a 42-48 Mdal plasmid (pYV), which encodes for many of the virulence-associated phenotypes (84). These include Ca2+-dependent growth, mouse lethality, cytotoxicity, Sereny reaction, production of V and W antigens, serum resistance, and production of outer membrane proteins (YOPs). The pYV plasmid of Y. enterocolitica was subcloned and the region encoding for HEp-2 cell cytotoxicity and Sereny reaction was identified and sequenced (91). A 24 base oligonucleotide probe, SP12, targeted to this region was shown to be specific for the virulence plasmid. The use of SP12 for detecting pathogenic Y. enterocolitica isolates in artificially inoculated foods was also evaluated (65).
For additional information on specific probes, contact the authors as follows:
At FDA Seafood Research Center, 22201 23rd Dr., S.E., Bothell, WA 98021:
At FDA, 200 C Street, S.W., Washington, DC 20204:
In the future it may be possible to carry out DNA probe tests with one standardized condition. Unfortunately, the inherent differences in the length and sequence composition of oligonucleotide probes and the variable susceptibility of microorganisms to lysing conditions require the use of several buffers and hybridization and washing temperatures. The types of microorganisms to be tested dictate the media and growth conditions. To minimize any confusion about similar but slightly different conditions to be used with various gene probes, each procedure is listed separately in this chapter. Although this results in much repetition, each protocol is complete and can be followed step by step. However, it is recommended that all the method sections be read, since some concepts and techniques discussed in the context of a particular probe may be applied in the use of others. In addition, for reference, a procedure for the end-labeling of oligonucleotides is presented.
Four different techniques can be used for colony hybridization tests:
The first three techniques differ as to when solid media are inoculated. In the first, aliquots of the homogenized sample are plated immediately after blending. In the second, plates are inoculated after aliquots of the homogenized sample have been incubated under selective conditions. Samples from the first and second techniques are plated onto selective agar media whenever such appropriate media are available. For the third technique, individual positive colonies are re-streaked and an additional colony hybridization test is conducted to ensure that the initial positive or negative results can be repeated. With the last technique, a pure culture can be obtained without selective enrichment, and additional microbiological tests requiring a pure culture can then be performed.
Control cultures
Strains that are positive or negative for the various probe tests must be properly stored and periodically tested for the appropriate phenotypic characteristics. A test methodology other than a gene probe must be used to independently verify the genotype of the control microorganisms. Control cultures must also be stored appropriately to minimize the possibility of genetic change. Usually, freezing liquid cultures at -70°C in 10-25% glycerol will suffice, except for Vibrio species, which are particularly sensitive to cold. Appropriate control strains have been listed, but other strains can be used if they have been properly characterized.
Preparation of controls
The importance of running controls cannot be overemphasized. Perhaps the most rigorous (and time-consuming) procedure for preparing controls is the inoculation of a food sample with a known number of positive (or negative) control cells. The food sample should have been previously tested by conventional microbiological techniques and shown to be free of the pathogen currently being sought. To prepare controls, streak out positive and negative cultures (or spot them in an array if pure cultures are being tested) onto the same medium as that used for food samples. Process the controls and the sample unknowns in an identical fashion at the same time. Such controls will signal if the cell growth, lysis, and hybridization steps have been successful. Although not serving as filter preparation controls, filter test strips will control for hybridization conditions. They can be prepared in advance by spotting control cultures into a repeated array. After cell lysis, cut the support into strips and add a strip to the hybridization mixture to control for conditions.
PROCEDURES
Enterotoxigenic Escherichia coli: Heat-Stable Enterotoxin (Human), Heat-Stable Enterotoxin (Porcine), and Heat-Labile Enterotoxin
Growth
Filter preparation--NOTE: If pure cultures are to be sought by picking colonies from original master plate after "positive" areas are found, use sterile Whatman 541 filters. Wrap filters in aluminum foil and autoclave on liquid cycle.
Hybridization
(NOTE: Several filters may be processed in the same petri dish. See Kaysner et al. (48) for protocol.)
Washing
Autoradiography
Controls
These control strains must have been confirmed for enterotoxin production by an appropriate non-probe assay, such as the suckling mouse test for ST or the mouse Y1 adrenal cell test for LT. Because the genes for these toxins are usually found on plasmids, control strains should be stored frozen at -70°C in 10% glycerol.
The following strains are suitable for use as controls:
| Strain | Reaction | Reference |
|---|---|---|
| C600 (pEWD299) | LT+ | 8 |
| H10407 | STH+, STP+, LT+ | 110 |
| ATCC 25922 | - | |
| HB101 (pBR313) | - | |
| C600 (pBR322) | - |
Interpretation of results
Confirmation
Invasive Shigella Species and Escherichia coli
Growth--enumeration method
Growth--presence/absence method
Filter preparation--see NOTE under PROCEDURES, above.
To prepare filters using a microwave, use filter preparation method for E. coli SLT.
Hybridization
NOTE: Several filters may be processed in the same petri dish. See Kaysner et al. (48) for protocol.
Washing
Autoradiography--see procedure under Enterotoxigenic E. coli, above.
Controls
The following strains have been used successfully as controls for the inv gene probe:
| Strain | Reaction | Reference |
|---|---|---|
| S. flexneri M90T | Positive | 97 |
| S. flexneri M90T-55 | Negative | 97 |
| E. coli (EIEC) M41 63 | Positive | 32 |
| HB101 (pBR313) | Negative | 32 |
Interpretation of results
Isolation of suspected colonies
Match spots present on autoradiogram to area on plate from which filter was made. Pick colony and establish pure culture. Follow procedure described in Chapter 6 to isolate and identify any Shigella spp.
NOTE: Compositing procedures may be applied to this method (see Chapter 1, on sample handling).
The most difficult problem is deciding what dilutions are necessary for plating. Because the bacterial flora in each food varies, a fixed number for the dilutions would be inadequate. For foods with a low microbial flora, dilutions of 102, 103, and 104 are adequate, whereas foods with a high bacterial background, e.g., alfalfa sprouts, require higher dilutions (103-106). Duplicate plates should be made for each dilution.
Confirm any positive cultures by using the Congo Red assay (94).
Escherichia coli Shiga-Like Toxin (SLT)
Growth--standard procedure
Growth--low levels expected
If very low levels of EHEC are suspected, prepare filters from enriched food samples, although this will make it impossible to obtain quantitative results.
Growth--control strains and pure cultures for screening
NOTE: Store isolates in 10% glycerol at -70°C if possible.
Filter preparation--see NOTE under PROCEDURES, above.
Hybridization
NOTE: Several filters may be processed in the same petri dish; however, this method has not been tested with this probe. See Kaysner et al. (48) for protocol.
Washing
Autoradiography--see procedure given under Enterotoxigenic E. coli, above.
Controls
Store control strains (and other pure cultures to be tested) preferably at -70°C in 10% glycerol or on BHI slants at room temperature. The following strains have been tested for the presence of the SLT gene by cytotoxin-neutralization assays and by colony hybridization with gene probes.
| Strain | Reaction | Reference |
|---|---|---|
| C600 | Negative | 104 |
| O157:H7 (933) | SLT I and SLT II | 90 |
| C600 (933J) | SLT I | 81, 104 |
| O26:H11 (H30) | SLT I | 52, 80 |
Interpretation of results
Confirmation
Pick probe-positive colonies from master plates (which were stored at 4°C) and confirm as E. coli. Serotype these isolates (17,60) and test for production of SLT by cytotoxicity assay (27) as modified by O'Brien (80).
Perform toxin neutralization assay according to O'Brien and LaVeck (79).
Growth and filter preparation--see procedures under E. coli Shiga-like toxin (SLT).
Hybridization and washing--see procedures under Yersinia pseudotuberculosis INV-3, but use 60°C as the optimal washing temperature.
Autoradiography--see procedure under Enterotoxigenic E. coli, above.
Controls--Available from Peter Feng, Division of Microbiological Studies (HFS-516), FDA, 200 C Street, S.W., Washington, DC 20204.
| Strain | Reaction |
|---|---|
| EC260 (O157:H7) | Positive |
| EC258 (MUG [+] E. coli) | Negative |
| EC36 (MUG [-] E. coli) | Negative |
Listeria monocytogenes: Combination of Invasion-Associated
Protein (iap) and Hemolysin (hly) Gene Probes - AD713
Growth--enumeration method
Growth--pure cultures
Filter preparation
Hybridization
Washing
Autoradiography--see procedure given under Enterotoxigenic E. coli, above.
Controls: Use filters inoculated with L. monocytogenes and L. innocua.
| Strain | Reaction | Reference |
|---|---|---|
| L. monocytogenes Scott A | Positive | 25 |
| L. innocua ATCC 33090 | Negative | 25 |
Interpretation of results
Confirm positive cultures with BAM procedures recommended for Listeria.
Staphylococcus aureus entB
Growth
Filter preparation--see NOTE under PROCEDURES, above.
With this probe, nylon supports, such as Nylon 66 (Micron Separations Inc., Westborough, MA 01581), generate less nonspecific background than do Whatman 541 filters.
Hybridization
Washing
Autoradiography--see procedure given under Enterotoxigenic E. coli, above.
| Strain | Reaction |
|---|---|
| SA 49 (243) | Positive |
| SA 54 (D87) | Negative |
Interpretation of results
Confirm toxin production by using a rapid method, as listed in Appendix 1.
Vibrio cholerae ctxA11
Growth--pure cultures
Filter preparation--see NOTE under PROCEDURES, above.
Hybridization
NOTE: Several filters may be processed in the same petri dish. See Kaysner et al. (48) for protocol.
Washing
Autoradiography--see procedure given under Enterotoxigenic E. coli, above.
Controls
| Strain | Reaction | Reference |
|---|---|---|
| ATCC 14033 | Positive | 99 |
| E. coli H10407 | Negative (high stringency) | 99 |
| E. coli H10407 | Positive (low stringency) | 99 |
Interpretation of results: Compare intensity of control spots with sample spots. Confirm positive cultures with BAM procedures recommended for V. cholerae.
Vibrio parahaemolyticus tdh3
The colony hybridization protocol is identical to that of V. cholerae, including the 45°C washing temperature. Confirm by performing those tests required to identify V. parahaemolyticus.
| Strain | Reaction | Reference |
|---|---|---|
| WP1 | Positive | 75 |
| S162-71 | Negative | 75 |
| ATCC 17802 | Positive | 75 |
Plating medium is modified CPC agar (M98). The colony hybridization protocol is identical to that for V. cholerae, except that washing temperature is 60°C. Confirm by performing those tests required to identify V. vulnificus.
| Strain | Reaction |
|---|---|
| ATCC 27562 | Positive |
| ATCC 14033 | Negative |
Growth--contaminated foods
NOTE: If samples are suspected to be heavily contaminated with other microflora, this level may be reduced by brief treatment with alkali, as demonstrated by Jagow and Hill (42).
Growth--pure culture testing for invasiveness and pathogenicity
Filter preparation--see NOTE under PROCEDURES, above.
Hybridization
NOTE: Several filters may be processed in the same petri dish. See Kaysner et al. (48) for protocol.
Washing
Autoradiography--see procedure given under Enterotoxigenic E. coli, above.
Controls
| Strain | Reaction |
|---|---|
| Yp188 isolate 82-599 | Positive |
| Ye133 (8081) | Negative |
Interpretation of results
Yersinia enterocolitica Chromosomal Probe PF-13
The colony hybridization protocol is identical to that described for INV-3, except that the optimal wash temperature for this probe is 47°C.
Controls
| Strain | Reaction |
|---|---|
| Ye133 (8081) | Positive |
| Yp188 | Negative |
The colony hybridization protocol is identical to that described for INV-3,except that the optimal wash temperature for this probe is 57°C.
Controls
| Strain | Reaction |
|---|---|
| Ye133 (8081) | Positive |
| Yf225 (Y. fredericksenii) | Negative |
End-Labeling of Oligonucleotides
Acknowledgments
We thank Ashok Chopra, Department of Microbiology, University of Texas Medical Branch, Galveston, TX, for information on the use of probe ctxA11. Drs. Mary Trucksess and Kristina Williams, FDA, Washington, DC, kindly provided unpublished information on the probe for staphylococcal enterotoxin.
References
1. Bergdoll, M.S. 1979. Staphylococcal intoxications, pp. 443-493. In: Foodborne Infections and Intoxications. H. Riemann and F.L. Bryan (eds). Academic Press, New York.
2. Betley, M.J., and J.J. Mekalanos. 1988. Nucleotide sequence of the type A staphylococcal enterotoxin gene. J. Bacteriol. 170:34-41.
3. Bohach, G.A., and P.M. Schlievert. 1987. Nucleotide sequence of the staphylococcal enterotoxin C1 gene and relatedness to other pyrogenic toxins. Mol. Gen. Genet. 209:15-20.
4. Boileau, C.R., H.M. d'Hauteville, and P.J. Sansonetti. 1984. DNA hybridization technique to detect Shigella species and enteroinvasive Escherichia coli. J. Clin. Microbiol. 20:959-961.
5. Cossart, P., M.F. Vicente, J. Mengaud, F. Baquero, J.C. Perez-Diaz, and P. Berche. 1989. Listeriolysin O is essential for virulence of Listeria monocytogenes: direct evidence obtained by gene complementation. Infect. Immun. 57:3629-3636.
6. Couch, J.L., M.T. Soltis, and M.J. Betley. 1988. Cloning and nucleotide sequence of the type E staphylococcal enterotoxin gene. J. Bacteriol. 170:2954-2960.
7. Curiale, M.S., M.J. Klatt, and M.A. Mozola. 1990. Colorimetric deoxyribonucleic acid hybridization assay for rapid screening of Salmonella in food: collaborative study. J. Assoc. Off. Anal. Chem. 73:248-256.
8. Dallas, W.S., D.M. Gill, and S. Falkow. 1979. Cistrons encoding Escherichia coli heat-labile toxin. J. Bacteriol. 139:850-858.
9. Datta, A.R., and B.A. Wentz. 1989. Identification and enumeration of virulent Listeria strains. Int. J. Food Microbiol. 8:259-264.
10. Datta, A.R., B. A. Wentz, D. Shook, and M.W. Trucksess. 1988. Synthetic oligodeoxyribonucleotide probes for detection of Listeria monocytogenes. Appl. Environ. Microbiol. 54:2933-2937.
11. Datta, A.R., B.A. Wentz, and J. Russell. 1990. Cloning of the listeriolysin O gene and development of specific gene probes for Listeria monocytogenes. Appl. Environ. Microbiol. 56:3874-3877.
12. Dean, A.G., Y. Chiang, R.G. Williams, and L.B. Handen. 1972. Test for Escherichia coli enterotoxin using infant mice: application in a study of diarrhea in children in Honolulu. J. Infect. Dis. 125:407-411.
13. Donta, S.T., M. King, and K. Sloper. 1973. Induction of steroido-genesis in tissue culture by cholerae enterotoxin. Nature New Biol. 243:246-247.
14. Doyle, M.P., and J.L. Schoeni. 1987. Isolation of Escherichia coli O157:H7 from retail fresh meats and poultry. Appl. Environ. Microbiol. 53:2394-2396.
15. Dupont, H.L., S.B. Formal, R.B. Hornick, M.J. Snyder, J.P. Libonati, D.G. Sheahan, E.H. LaBrec, and J.P. Kalas. 1971. Pathogenesis of Escherichia coli. N. Engl. J. Med. 285:1-9.
16. Erlich, H.A., R. Gibbs, and H.H. Kazazian, Jr., Eds. 1989. Polymerase Chain Reaction. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
17. Ewing, W.H. 1986. Edward and Ewing's Identification of Enterobacteriaceae, 4th ed. Elsevier, New York.
18. Feinberg, A.P., and B. Vogelstein. 1983. A technique for radiolabeling DNA restriction endonuclease fragments to high specific activity. Anal. Biochem. 132:6-13.
19. Feng, P. 1989. Oligonucleotide probes specific for Yersinia pseudotuberculosis. ASM Abstracts, p. 321.
20. Feng, P. 1992. Identification of invasive Yersinia spp. using oligonucleotide probes. Mol. Cell. Probes 6:291-297.
21. Feng, P., R. Lum, and G. Chang. 1991. Identification of uidA gene sequences in beta-D-glucuronidase (-) Escherichia coli. Appl. Environ. Microbiol. 57:320-323.
22. Field, M., L. Graf, W. Laird, and P. Smith. 1978. Heat stable enterotoxin of Escherichia coli: in vitro effects on guanylate cyclase activity, cyclic GMP concentration and ion transport in small intestine. Proc. Natl. Acad. Sci. USA 75:2800-2804.
23. Fitts, R., M. Diamond, C. Hamilton, and M. Neri. 1983. DNA-DNA hybridization assay for detection of Salmonella spp. in food. Appl. Environ. Microbiol. 46:1146-1151.
24. Flamm, R.K., D.J. Himrichs, and M.F. Thomashow. 1989. Cloning of the gene encoding a major secreted polypeptide of Listeria monocytogenes and its potential use as a species specific probe. Appl. Environ. Microbiol. 55:2251-2256.
25. Fleming, D.W., S.L. Cochi, K.L. MacDonald, J. Brondum, P.S. Hayes, B.D. Plikaytis, M.B. Holmes, A. Audurier, C.V. Broome, and A.L. Reingold. 1985. Pasteurized milk as a vehicle of infection in an outbreak of listeriosis. N. Engl. J. Med. 312:404-407.
26. Gaillard, J.L., P. Berche, and P. Sansonetti. 1986. Transposon mutagenesis as a tool to study the role of hemolysin in the virulence of Listeria monocytogenes. Infect. Immun. 52:50-55.
27. Gentry, M.K., and J.M. Dalrymple. 1980. Quantitative microtiter cytotoxicity assay for Shigella toxin. J. Clin. Microbiol. 12:361-366.
28. Gray, L.D., and A.S. Kreger. 1985. Purification and characterization of an extracellular cytolysin produced by Vibrio vulnificus. Infect. Immun. 48:62-72.
29. Grunstein, M., and D.S. Hogness. 1975. Colony hybridization: method for the isolation of cloned DNAs that contain a specific gene. Proc. Natl. Acad. Sci. USA 72:3961-3965.
30. Guerrant, R.L., L.L. Brunton, T. Schnaitman, L.I. Rebhun, and A.G. Gilman. 1974. Cyclic adenosine monophosphate and alteration of Chinese hamster ovary cell morphology: a rapid, sensitive in vitro assay for the enterotoxins of Vibrio cholerae and Escherichia coli. Infect. Immun. 10:320-327.
31. Gyles, C., and D.A. Barnum. 1969. A heat labile enterotoxin from strains of Escherichia coli enteropathogenic for pigs. J. Infect. Dis. 120:419-426.
32. Hale, T.L., P.J. Sansonetti, P.A. Schad, S. Austin, and S.B. Formal. 1983. Characterization of virulence plasmid and plasmid-associated outer membrane proteins in Shigella flexneri, Shigella sonnei, and Escherichia coli. Infect. Immun. 40:340-350.
33. Hanchalay, S., J. Seriwatana, P. Echeverria, J. Homgren, C. Tirapat, S.L. Moseley, and D.N. Taylor. 1985. Non-Ol Vibrio cholerae in Thailand: homology with cloned cholera toxin genes. J. Clin. Microbiol. 21:288-289.
34. Heisick, J.E., F.M. Harrell, E.H. Peterson, S. McLaughlin, D.E. Wagner, I.V. Wesley, and J. Brymer. 1989. Comparison of four procedures to detect Listeria spp. in foods. J. Food Prot. 52:154-157.
35. Hill, W.E., W.L. Payne, G. Zon, and S.L. Moseley. 1985. Synthetic oligodeoxyribonucleotide probes for detecting heat-stable enterotoxin-producing Escherichia coli by DNA colony hybridization. Appl. Environ. Microbiol. 50:1187-1191.
36. Hill, W.E., B.A. Wentz, W.L. Payne, J.A. Jagow, and G. Zon. 1986. DNA colony hybridization method using synthetic oligonucleotides to detect enterotoxigenic Escherichia coli: collaborative study. J. Assoc. Off. Anal. Chem. 69:531-536.
37. Hill, W.E., S.P. Keasler, M.W. Trucksess, P. Feng, C.A. Kaysner, and K.A. Lampel. 1991. Polymerase chain reaction identification of Vibrio vulnificus in artificially contaminated oysters. Appl. Environ. Microbiol. 57:707-711.
38. Isberg, R., and S. Falkow. 1985. A single genetic locus encoded by Yersinia pseudotuberculosis permits invasion of cultured animal cells by Escherichia coli K-12. Nature 317:262-264.
39. Isberg, R., D.L. Voorhis, and S. Falkow. 1987. Identification of invasion: a protein that allows enteric bacteria to penetrate cultured mammalian cells. Cell 50:769-778.
40. Jablonski, E., E.W. Moomaw, R.H. Tullis, and J.L. Ruth. 1986. Preparation of oligodeoxynucleotide--alkaline phosphatase conjugates and their use of hybridization probes. Nucleic Acids Res. 14:6115-6128.
41. Jackson, M.P., R.J. Neill, A.D. O'Brien, R.K. Holmes, and J.W. Newland. 1987. Nucleotide sequence analysis and comparison of the structural genes for Shiga-like toxin I and Shiga-like toxin II encoded by bacteriophages from Escherichia coli 933. FEMS Microbiol. Lett. 44:109-ll4.
42. Jagow, J.A., and W.E. Hill. 1988. Enumeration of virulent Yersinia enterocolitica colonies by DNA colony hybridization using alkaline treatment and paper filter. Mol. Cell. Probes 2:189-195.
43. Jones, C.L., and S.A. Kahn. 1986. Nucleotide sequence of the enterotoxin B gene from Staphylococcus aureus. J. Bacteriol. 166:29-33.
44. Joseph, S.W., R.R. Colwell, and J.B. Kaper. 1982. Vibrio parahaemolyticus and related halophilic vibrios. Crit. Rev. Microbiol. 10:77-124.
45. Kaper, J.B., R.K. Campen, R.J. Seidler, M.M. Baldini, and S. Falkow. 1984. Cloning of the thermostable direct or Kanagawa phenomenon-associated hemolysin of Vibrio parahaemolyticus. Infect. Immun. 45:290-292.
46. Karmali, M.A. 1989. Infection by verocytotoxin producing Escherichia coli. Clin. Microbiol. Rev. 2:15-18.
47. Kathariou, S., P. Metz, H. Hof, and W. Goebel. 1987. Tn916 induced mutations in the hemolysin determinant affecting virulence of Listeria monocytogenes. J. Bacteriol. 169:1291-1297.
48. Kaysner, C.A., S.D. Weagant, and W.E. Hill. 1988. Modification of the DNA colony hybridization technique for multiple filter analysis. Mol. Cell. Probes 2:255-260.
49. Keller, G.H., and M.M. Manak. 1989. DNA probes. Stockton Press, New York.
50. King, W., S. Raposa, J. Warshaw, A. Johnson, D. Halbert, and J.D. Klinger. 1989. A new colorimetric nucleic acid hybridization assay for Listeria in foods. Int. J. Food Microbiol. 8:226-232.
51. Kohler, S., M. Leimeister-Wachter, T. Chakraborty, F. Lottspeich, and W. Goebel. 1990. The gene coding for protein p60 of Listeria monocytogenes
and its use as a specific probe for Listeria monocytogenes. Infect. Immun. 58:1943-1950.
52. Konowalchuk, J., J.I. Spiers, and S. Stavric. 1977. Vero response to a cytotoxin of Escherichia coli. Infect. Immun. 18:775-779.
53. Kuhn, M., and W. Goebel. 1989. Identification of an extracellular protein of Listeria monocytogenes possibly involved in intracellular uptake by mammalian cells. Infect. Immun. 57:55-61.
54. Lampel, K.A., J.A. Jagow, M.L. Troxell, S. Tucker, and A.T. Maurelli. 1995. An oligonucleotide probe for the detection of enteroinvasive Escherichia coli and Shigella spp. in foods. (Submitted for publication.)
55. Leimeister-Wachter, M., and T. Chakraborty. 1989. Detection of listeriolysin, the thiol dependent hemolysin in Listeria monocytogenes, Listeria ivanovii and Listeria seeligeri. Infect. Immun. 57:2350-2357.
56. Leong, J., A.C. Vinal, and W.S. Dallas. 1985. Nucleotide sequence comparison between heat-labile toxin B-subunit cistrons from Escherichia coli of human and porcine origin. Infect. Immun. 48:73-77.
57. Lockman, H., and J.B. Kaper. 1983. Nucleotide sequence analysis of the A2 and B subunits of Vibrio cholerae enterotoxin. J. Biol. Chem. 258:722-13726.
58. Lockman, H.A., J.E. Galen, and J.B. Kaper. 1984. Vibrio cholerae enterotoxin genes: nucleotide sequence analysis of DNA encoding ADP-ribosyltransferase. J. Bacteriol. 159:1086-1089.
59. Maas, R. 1983. An improved colony hybridization method with significantly increased sensitivity for detection of single genes. Plasmid 10:296-298.
60. Marques, L.R.M., M.A. Moore, J.G. Wells, I.K. Wachsmuth, and A.D. O'Brien. 1986. Production of Shiga-like toxin by Escherichia coli. J. Infect. Dis. 154:338-341.
61. Maurelli, A.T., B. Baudry, H. d'Hauteville, T.L. Hale, and P.J. Sansonetti. 1985. Cloning of plasmid DNA sequences involved in invasion of HeLa cells by Shigella flexneri. Infect. Immun. 49:164-171.
62. Meinkoth, J., and G. Wahl. 1984. Hybridization of nucleic acids immunobilized on solid supports. Anal. Biochem. 138:267-284.
63. Mekalanos, J.J., D.J. Swartz, G.D.N. Pearson, N. Hardford, F. Groyne, and M. deWilde. 1983. Cholera toxin genes: nucleotide sequence, deletion analysis and vaccine development. Nature 306:551-557.
64. Mengaud, J., M. Vicente, J. Chemevert, J.M. Pereira, C. Geoffroy, B. Gicquel-Sanzey, F. Baquero, J. Perez-Diaz, and P. Cossart. 1988. Expression in Escherichia coli and sequence analysis of the listeriolysin O determinant of Listeria monocytogenes. Infect. Immun. 56:766-772.
65. Miliotis, M.D., J.E. Galen, J.B. Kaper, and J.G. Morris. 1989. Development and testing of a synthetic oligonucleotide probe for the detection of pathogenic Yersinia strains. J. Clin. Microbiol. 27:1667-1670.
66. Miller, V.A., and S. Falkow. 1988. Evidence of two genetic loci in Yersinia enterocolitica that can promote invasion of epithelial cells. Infect. Immun. 56:1242-1248.
67. Miller, V., J.J. Farmer III, W.E. Hill, and S. Falkow. 1989. The ail locus is found uniquely in Yersinia enterocolitica serotypes commonly associated with disease. Infect. Immun. 57:121-131.
68. Miller, V.L., J.B. Bliska, and S. Falkow. 1990. Nucleotide sequence of the Yersinia enterocolitica ail gene and characterization of the ail protein product. J. Bacteriol. 172:1062-1069.
69. Miyamoto, Y., Y. Obara, T. Nikkawa, S. Yamai, T. Kato, Y. Yamada, and M. Ohashi. 1980. Simplified purification and biophysicochemical characteristics of Kanagawa phenomenon-associated hemolysin of Vibrio parahaemolyticus. Infect. Immun. 28:567-576.
70. Mohamood, A., B. Wentz, A. Datta, and B. Eribo. 1990. Application of a synthetic listeriolysin O gene probe to the identification of hemolytic Listeria monocytogenes in retail ground beef. ASM Abstracts, p. 279.
71. Morris, J.G., Jr., and R.E. Black. 1985. Cholera and other vibrioses in the United States. N. Engl. J. Med. 312:343-350.
72. Morris, J.G., Jr., A.C. Wright, D.M. Roberts, P.K. Wood, L.M. Simpson, and J.D. Oliver. 1987. Identification of environmental Vibrio vulnificus isolates with a DNA probe for the cytotoxin-hemolysin gene. Appl. Environ. Microbiol. 53:193-195.
73. Moseley, S.L., and S. Falkow. 1980. Nucleotide sequence homology between the heat-labile enterotoxin gene of Escherichia coli and Vibrio cholerae deoxyribonucleic acid. J. Bacteriol. 144:444-446.
74. Moseley, S.L., J.W. Hardy, M.I. Huq, P. Echeverria, and S. Falkow. 1983. Isolation and nucleotide sequence determination of a gene encoding a heat-stable enterotoxin of Escherichia coli. Infect. Immun. 39:1167-1174.
75. Nishibuchi, M., and J.B. Kaper. 1985. Nucleotide sequence of the thermostable direct hemolysin gene of Vibrio parahaemolyticus. J. Bacteriol. 162:558-564.
76. Nishibuchi, M., M. Ishibashi, Y. Takeda, and J.B. Raper. 1985. Detection of the thermostable direct hemolysin gene and related DNA sequences in Vibrio parahaemolyticus and other Vibrio species by the DNA colony hybridization test. Infect. Immun. 49:481-486.
77. Nishibuchi, M., W.E. Hill, G. Zon, W.L. Payne, and J.B. Kaper. 1986. Synthetic oligonucleotide probes to detect Kanagawa phenomenon-positive Vibrio parahaemolyticus. J. Clin. Microbiol. 23:1091-1095.
78. Notermans, S., K.J. Heuvelman, and K. Wernars. 1988. Synthetic enterotoxin B DNA probes for detection of enterotoxigenic Staphylococcus aureus strains. Appl. Environ. Microbiol. 54:531-533.
79. O'Brien, A.D., and G.D. LaVeck. 1983. Purification and characterization of a Shigella dysenteriae I-like toxin produced by Escherichia coli. Infect. Immun. 40:675-683.
80. O'Brien, A.D., G.D. LaVeck, M.R. Thompson, and S.B. Formal. 1982. Production of Shigella dysenteriae type I like cytotoxin by Escherichia coli. J. Infect. Dis. 146:763-769.
81. O'Brien, A.D., J.W. Newland, S.F. Miller, R.K. Holmes, H.W. Smith, and S.B. Formal. 1984. Shiga-like toxin converting phages from E. coli strains that cause hemorrhagic colitis and infantile diarrhea. Science 226:694-696.
82. Picken, R.N., A.J. Mazaitis, W.K. Maas, M. Rey, and H. Heyneker. 1983. Nucleotide sequence of the gene for heat-stable enterotoxin II of Escherichia coli. Infect. Immun. 42:269-275.
83. Picken, R.N., Z. Wang, and H.L. Yang. 1987. Molecular cloning of species-specific DNA probe for Campylobacter jejuni. Mol. Cell. Probes 1:245-259.
84. Portnoy, D.A., and R.J. Martinez. 1985. Role of plasmids in the pathogenicity of Yersinia species. Curr. Top. Microbiol. Immunol. 11: 29-51.
85. Ranelli, D.M., C.L. Jones, M.B. Johns, G.J. Mussey, and S.A. Kahn. 1985. Molecular cloning of staphylococcal enterotoxin B gene in Escherichia coli and Staphylococcus aureus. Proc. Natl. Acad. Sci. USA 82:5850-5854.
86. Rashtchian, A., M.A. Abbott, and M. Shaffer. 1987. Cloning and characterization of genes coding for ribosomal RNA in Campylobacter jejuni. Curr. Microbiol. 14:311-317.
87. Rashtchian, A., J. Eldredge, M. Ottaviani, M. Abbott, G. Mock, D. Lovern, J. Klinger, and G. Parsons. 1987. Immunological capture of nucleic acid hybrids and application to nonradioactive DNA probe assay. Clin. Chem. 33:1526-1530.
88. Richardson, C.C. 1971. Polynucleotide kinase from Escherichia coli infected with bacteriophage T4, p. 815. In: Procedures in Nucleic Acid Research, Vol. 2, p. 815. G.L. Cantoni and D.R. Davies (eds). Harper and Row, New York.
89. Rigby, P.W.J., M. Dieckmann, C. Rhodes, and P. Berg. 1977. Labeling deoxyribonucleic acid to high specific activity in vitro by nick translation with DNA polymerase I. J. Mol. Biol. 113:237-251.
90. Riley, L.W., R.S. Temis, and S.D. Helgerson. 1983. Hemorrhagic colitis associated with a rare Escherichia coli serotype. N. Engl. J. Med. 308:681-685.
91. Robins-Brown, R., M.D. Miliotis, S. Cianciosi, V.L. Miller, S. Falkow, and J.G. Morris. 1989. Evaluation of DNA colony hybridization and other techniques for detection of virulence in Yersinia species. J. Clin. Microbiol. 27:644-650.
92. Saiki, R.K., S. Scharf, F. Faloona, K.B. Mullis, G.T. Horn, and H.A. Erlich. 1985. Enzymatic amplification of beta-globin genomic sequence and restriction analysis for diagnosis of sickle cell anemia. Science 230:1350-1354.
93. Saiki, R.K., D.H. Gelfand, S. Stoffel, S.J. Scharf, R. Higuchi, G.T. Horn, K.B. Mullis, and H.A. Erlich. 1988. Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science 239:487-491.
94. Sakai, T., C. Sasakawa, S. Makino, K. Kamata, and M. Yoshikawa. 1986. Molecular cloning of a genetic determinant for Congo Red binding ability which is essential for the virulence of Shigella flexneri. Infect. Immun. 51:476-482.
95. Sakurai, J., A. Matsuzaki, and T. Miwatani. 1973. Purification and characterization of thermostable direct hemolysin of Vibrio parahaemolyticus. Infect. Immun. 8:775-780.
96. Sansonetti, P.J., D.J. Kopecko, and S.B. Formal. 1981. Shigella soneii plasmids: evidence that a large plasmid is necessary for virulence. Infect. Immun. 34:75-83.
97. Sansonetti, P.J., D.J. Kopecko, and S.B. Formal. 1982. Involvement of a plasmid in the invasive ability of Shigella flexneri. Infect. Immun. 35:852-860.
98. Sereny, B. 1955. Experimental shigella keratoconjunctivitis. Acta Microbiol. Acad. Sci. Hung. 2:293-296.
99. Skerman, F.J., S.B. Formal, and S. Falkow. 1972. Plasmid-associated enterotoxin production in a strain of Escherichia coli isolated from humans. Infect. Immun. 5:622-624.
100. Small, P.L.C., and S. Falkow. 1988. Identification of regions on a 230 kilobase plasmid from enteroinvasive Escherichia coli that are required for entry into HEp-2 cells. Infect. Immun. 56:225-229.
101. So, M., and B.J. McCarthy. 1980. Nucleotide sequences of bacterial transposon TN 1681 encoding a heat stable toxin (ST) and its identification in enterotoxigenic Escherichia coli strains. Proc. Natl. Acad. Sci. USA 77:4011-4015.
102. So, M., W. Dallas, and S. Falkow. 1978. Characterization of an Escherichia coli plasmid encoding for synthesis of heat-labile toxin: molecular cloning of the toxin determinant. Infect. Immun. 21:405-411.
103. Spicer, E.K., and J.A. Noble. 1982. Escherichia coli heat-labile enterotoxin: nucleotide sequence of the A subunit gene. J. Biol. Chem. 257:5716-5721.
104. Strockbine, N.A., L.R.M. Marques, J.W. Newland, H.W. Smith, R.K. Holmes, and A.D. O'Brien. 1986. Two toxin converting phages from Escherichia coli O157:H7 strain 933 encode antigenically distinct toxins with similar biological activities. Infect. Immun. 53:135-140.
105. Szabo, R.A., E.C.D. Todd, and A. Jean. 1986. Method to isolate Escherichia coli O157:H7 from food. J. Food Prot. 49:768-772.
106. Taniguchi, H., H. Ohta., M. Ogawa, and Y. Mizuguchi. 1985. Cloning and expression in Escherichia coli of Vibrio parahaemolyticus thermostable direct hemolysin and thermolabile hemolysin genes. J. Bacteriol. 162:510-515.
107. Watson, J.D., N.H. Hopkins, J.W. Roberts, J.A. Steitz, and A.M. Weiner. 1987. Molecular Biology of the Gene, 4th ed., Benjamin/Cummings, Menlo Park, CA.
108. Wood, P.K., J.G. Morris, Jr., P.L.C. Small, O. Sethabutr, M.R.F. Toledo, L. Trabulsi, and J.B. Kaper. 1986. Comparison of DNA probes and the Sereny test for identification of invasive Shigella and Escherichia coli strains. J. Clin. Microbiol. 24:498-500.
109. Wright, A.C., J.G. Morris, Jr., D.R. Maneval, Jr., K. Richardson, and J.B. Kaper. 1985. Cloning of the cytotoxin-hemolysin gene of Vibrio vulnificus. Infect. Immun. 50:922-924.
110. Yamamoto, T., and T. Yokota. 1983. Sequence of heat-labile enterotoxin of Escherichia coli pathogenic for humans. J. Bacteriol. 155:728-733.
111. Yamamoto, T., T.A. Tamura, M. Ryoji, A. Kaji, T. Yokota, and T. Takano. 1982. Sequence analysis of the heat labile enterotoxin subunit B gene originating in human enterotoxigenic Escherichia coli. J. Bacteriol. 152:506-509.
112. Yamamoto, K., A.C. Wright, J.B. Kaper, and J.G. Morris, Jr. 1990. Cytolysin gene of V. vulnificus: sequence and relationship to V. cholerae El Tor hemolysin. Infect. Immun. 58:2706-2709.
Hypertext Source: Bacteriological Analytical Manual, 8th Edition, Revision A, 1998. Chapter 24.
Hypertext updated by kwg/cjm 2001-OCT-24