Red Cell Membrane Disorder Mutations Database
Hereditary Spherocytosis (
HS), Hereditary Elliptocytosis (
HE), and Hereditary Pyropoikilocytosis (
HPP) are inherited disorders of the erythrocyte membrane associated with hemolytic anemia [
1,
2]. These disorders are characterized by genetic heterogeneity [
3,
4] as well as clinical and laboratory heterogeneity. This database contains confirmed mutations associated with these diseases. We welcome additions and descriptions of new mutations. Information can be sent to either Dr. Patrick G. Gallagher of the Yale Center for Blood Disorders or Dr. David M. Bodine of NHGRI using the
submission form.
Hereditary Spherocytosis

Hereditary Spherocytosis (HS) takes its name from the spherical-shaped erythrocytes observed on the peripheral blood smear of affected patients (Right). Clinically, the "typical" HS patient presents with mild to moderate anemia. However, the severity of the anemia in HS patients varies widely, ranging from nearly asymptomatic to severe, transfusion-dependence. Other hematologic findings include an elevated number of reticulocytes, elevated mean corpuscular hemoglobin concentration, and increased erythrocyte osmotic fragility after incubation.
HS is seen in families of all races. It is the most common inherited anemia in individuals of northern European descent, affecting approximately 1 in 1000-2500 individuals. Most HS patients have a positive family history of the disease. Due to the wide range of clinical severity, many HS patients are diagnosed during the analysis of a more severely affected family member. Recent genetic studies have revealed that a large number of HS mutations are de novo.[5]
The genes mutated in HS patients include ANK1 (ankyrin 1, erythrocytic), SLC4A1 (solute carrier family 4, anion exchanger, member 1 (erythrocyte membrane protein band 3, Diego blood group)), SPTB (pectrin, beta, erythrocytic), SPTA1 (spectrin, alpha, erythrocytic 1 (elliptocytosis 2)), and EPB42 (erythrocyte membrane protein band 4.2)[5]. ANK1 mutations are the most common, followed by SLC4A1 and SPTB mutations. In most HS patients, inheritance is autosomal dominant. In general, HS mutations are private, i.e., each individual kindred has a unique mutation. Individual HS pedigrees have been identified with autosomal recessive inheritance of mutations in either SPTA1 or EPB42. Rare examples of "double dominant" HS due to defects in SLC4A1 or SPTB resulting in fetal death or severe transfusion-dependent hemolytic anemia presenting in the neonatal period have also been reported.
Hereditary Elliptocytosis and Hereditary Pyropoikilocytosis
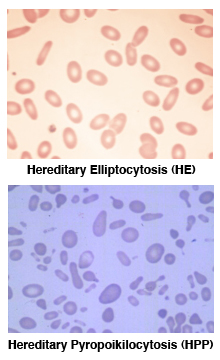
Like HS, Hereditary Elliptocytosis (HE) takes its name from the elliptical, cigar-shaped erythrocytes observed on the peripheral blood smear (see HE at right)[
6] Clinically, HE is characterized by mild to no anemia, but HE is a heterogeneous disorder, ranging from asymptomatic to severe, life-threatening anemia. Depending on the degree of anemia, HE patients may also have elevated levels of reticulocytes and increased erythrocyte osmotic fragility after incubation, although most “typical” HE patients are asymptomatic.
In contrast, Hereditary Pyropoikilocytosis (HPP) is a more severe hemolytic anemia characterized by the presence of large numbers of non-uniform, fragmented erythrocytes on the peripheral smear (see HPP at right) with elevated levels of reticulocytes, microcytosis, and markedly abnormal erythrocyte osmotic fragility after incubation. HE and HPP are related disorders. Many HPP patients experience severe hemolytic anemia in childhood that may evolve into a milder HE as they get older. In HPP kindreds, up to a third of family members exhibit the phenotype of HE. [6,7]
HE is common in individuals of African and Mediterranean descent. It has been postulated that elliptocytes confer resistance to malaria. Worldwide, the incidence of HE is estimated at 1:2000-4000, approaching 1:100 in parts of Africa. The actual incidence is unknown, as most patients are asymptomatic. While most HE patients are diagnosed incidentally during testing for unrelated conditions, a positive family history is part of the diagnosis of HPP. Often, severely affected HE patients have been shown to have inherited asymptomatic HE mutations from both parents. Likewise, many HPP patients have been found to be homozygotes or compound heterozygotes for HE mutations.[7]
HE is inherited in an autosomal dominant pattern with rare cases of de novo mutations.[5,6] The genes mutated in HE/HPP are SPTA1 (alpha-spectrin), SPTB (beta-spectrin) and EPB41 (band 4.1), with SPTA1 mutations being the most common. The majority of the mutations alter the self-association regions of the spectrin proteins, leading to red cell membrane alterations.[6,8]




