


Medical Devices
Draft Guidance for Industry and Food and Drug Administration Staff - De Novo Classification Process (Evaluation of Automatic Class III Designation)
The Food and Drug Administration Safety and Innovation Act (FDASIA), signed into law on July 9, 2012, amended, among other sections, section 513(f) of the Federal Food, Drug, and Cosmetic Act. The guidance entitled New Section 513(f)(2) - Evaluation of Automatic Class III Designation, Guidance for Industry and CDRH Staff, issued on February 19, 1998, and the draft guidance entitled Draft Guidance for Industry and Food and Drug Administration Staff – De Novo Classification Process (Evaluation of Automatic Class III Designation), issued on October 3, 2011, were developed and issued prior to the enactment of FDASIA, and certain sections of these guidances may no longer be current as a result of FDASIA. The Center for Devices and Radiological Health is currently working on a new draft de novo guidance, that when finalized, will represent the FDA’s current thinking on this topic. Until the new draft de novo guidance is published, please contact 510(k) Staff at 301-796-5640 for information regarding the de novo process.
 DRAFT GUIDANCE
DRAFT GUIDANCE
This guidance document is being distributed for comment purposes only.
Document issued on: October 3, 2011
You should submit comments and suggestions regarding this draft document within 90 days of publication in the Federal Register of the notice announcing the availability of the draft guidance. Submit written comments to the Division of Dockets Management (HFA-305), Food and Drug Administration, 5630 Fishers Lane, rm. 1061, Rockville, MD 20852. Submit electronic comments to http://www.regulations.gov. Identify all comments with the docket number listed in the notice of availability that publishes in the Federal Register.
For questions regarding this document, contact Melissa Burns, 301-796-5616, melissa.burns@fda.hhs.gov or CBER’s Office of Communication, Outreach and Development at 1-800-835-4709 or 301-827-1800.
When final, this document will supersede “New Section 513(f)(2) - Evaluation of Automatic Class III Designation, Guidance for Industry and CDRH Staff” dated February 19, 1998.
 | U.S. Department of Health and Human Services Center for Biologics Evaluation and Research |
Preface
Additional Copies
Additional copies are available from the Internet. You may also send an e-mail request to dsmica@fda.hhs.gov to receive an electronic copy of the guidance or send a fax request to 301-847-8149 to receive a hard copy. Please use the document number (1760) to identify the guidance you are requesting.
Or contact:
Office of Communication, Outreach and Development, HFM-40
Center for Biologics Evaluation and Research
Food and Drug Administration
1401 Rockville Pike, Suite 200N, Rockville, MD 20852-1448
Internet:
http://www.fda.gov/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/default.htm
Tel: 800-835-4709 or 301-827-1800
E-mail: ocod@fda.hhs.gov
Draft Guidance for Industry and Food and Drug Administration Staff
De Novo Classification Process (Evaluation of Automatic Class III Designation)
This draft guidance, when finalized, will represent the Food and Drug Administration's (FDA's) current thinking on this topic. It does not create or confer any rights for or on any person and does not operate to bind FDA or the public. You can use an alternative approach if the approach satisfies the requirements of the applicable statutes and regulations. If you want to discuss an alternative approach, contact the FDA staff responsible for implementing this guidance. If you cannot identify the appropriate FDA staff, call the appropriate number listed on the title page of this guidance.
1. Introduction
The purpose of this document is to provide guidance on the process for the submission and review of petitions under section 513(f)(2) of the Federal Food, Drug, and Cosmetic Act (the FD&C Act), also known as the de novo classification process. This process provides a route to market for medical devices that are low to moderate risk, but that have been classified in class III because FDA has found them to be “not substantially equivalent” (NSE) to legally marketed predicate devices.
FDA's guidance documents, including this guidance, do not establish legally enforceable responsibilities. Instead, guidances describe the Agency's current thinking on a topic and should be viewed only as recommendations, unless specific regulatory or statutory requirements are cited. The use of the word should in Agency guidances means that something is suggested or recommended, but not required.
Throughout this guidance document, the terms “we,” “us” and “our” refer to FDA staff from the Center for Devices and Radiological Health (CDRH) or the Center for Biologics Evaluation and Research (CBER) involved in the review and decision-making aspects of the de novo classification process. “You” and “your” refers to the submitter of a de novo petition and/or related materials.
2. Background
A device may be classified in class III and require premarket approval via several different regulatory vehicles. In accordance with section 513(a)(1)(C) of the FD&C Act, FDA may promulgate a regulation which classifies a device type into class III based on the risks posed by the device and the inability of general and special controls to provide reasonable assurance of the safety and effectiveness of the device. All particular devices within this type are considered to be in class III and such devices are not eligible for the de novo classification process.
Alternatively, devices of a new type that FDA has not previously classified based on risk are “automatically” or “statutorily” classified into class III by operation of section 513(f)(1) of the FD&C Act, regardless of the level of risk they pose. This is because, by definition, a new type of device would not be within a type that was on the market before the 1976 Medical Device Amendments or that has since been classified into class I or class II.
This second scenario is what was targeted by Congress when it enacted section 513(f)(2) of the FD&C Act as part of the Food and Drug Administration Modernization Act of 1997 (FDAMA). The process created by this provision, which is referred to in FDAMA as the Evaluation of Automatic Class III Designation, will be referred to as the “de novo process”1 throughout this guidance document. Congress included this section to limit unnecessary expenditure of FDA and industry resources that could occur if lower risk devices were subject to premarket approval (PMA) under section 515 of the FD&C Act.
Although FDA has reviewed and granted a number of de novo petitions since the enactment of FDAMA, we believe the program has been under-utilized because of process inefficiencies. FDA believes that the process could be improved and greater clarity could be provided regarding suitability and data needed so that the de novo process may be a more viable pathway for novel low to moderate risk devices. Accordingly, FDA is issuing this draft guidance to provide updated recommendations for more efficient interactions with FDA, including what information to submit when seeking a path to market via the de novo process. This guidance describes a mechanism to provide (1) greater clarity about the suitability of a device for the de novo process, and (2) timely input on the type of data necessary to support de novo classification of a suitable device. When final, this guidance will replace “New Section 513(f)(2) – Evaluation of Automatic Class III Designation, Guidance for Industry and CDRH Staff,” dated February 19, 1998.
3. The De Novo Process
A novel type of device may be eligible for the de novo process if it has received an NSE determination as a result of a 510(k) submission. In this case, in accordance with section 513(f)(2), the submitter of a 510(k) may, within thirty (30) days of receipt of an NSE determination for that 510(k), submit a de novo petition requesting FDA to make a risk-based classification determination for the device under section 513(a)(1) of the FD&C Act. The de novo petition must include a description of the device and detailed information and reasons for any recommended classification. FDA must make a classification determination for the device that is the subject of the petition by written order within sixty (60) days of the request.
If we grant the de novo petition, the device is reclassified from class III into class I or class II. The device may then be marketed immediately and serve as a predicate device. Thereafter, we will also publish a notice in the Federal Register announcing the classification, the accompanying regulation, and the controls necessary to provide reasonable assurance of safety and effectiveness. If the petition is denied, the device remains in class III and may not be marketed.
3.1 When the De Novo Process May Be Used
FDA reviews de novo petitions for new2 devices that meet two threshold criteria. The first is that the new device is not within a device type that has been classified based on risk. The second is that the new device is statutorily classified into class III and FDA has provided “written notice” of this, i.e., an NSE determination in response to a 510(k) submission, within the last 30 days.
FDA will consider a de novo petition if the new device has been determined to be NSE due to: (1) the lack of an identifiable predicate device, (2) new intended use, or (3) different technological characteristics that raise new questions of safety and effectiveness. New devices that have been found to be NSE due to lack of performance data would generally be ineligible for the de novo process because lack of performance data means that a predicate device likely exists, so the device type likely has been classified. Similarly, if the new device is within a type for which there is an existing Class III classification regulation or an approved PMA, then the new device would not be eligible for de novo since it would be of a type that has been previously classified.
In addition, the following additional criteria should be met for a new device for which a de novo petition is submitted:
- The new device should be low to moderate risk and likely to meet the statutory standards for classification into class I or class II under section 513(a)(1) of the FD&C Act, e.g., general and/or special controls would provide reasonable assurance of the safety and effectiveness of the device; and
- You should sufficiently understand and be able to explain all of the risks and benefits of the new device such that all risks can be effectively mitigated through the application of general and/or special controls.
3.2 Submitting De Novo Information for FDA Review
This guidance outlines the two pathways for the de novo process:
- a pathway that is initiated with a “pre de novo submission” (PDS); or
- a pathway that is initiated with a 510(k) submission.
A PDS affords you early interaction with us regarding a new device for which you believe there is no predicate device and which you believe is low to moderate risk. For new devices that appear to be suitable for de novo, the PDS pathway also affords you the ability to follow the PDS with a concurrent 510(k) and de novo petition. The purpose of the PDS is for us to analyze whether a new device appears to be suitable for the de novo process, and, if so, to provide an opportunity to advise you on the documentation needed in the subsequent 510(k) and de novo petition. The primary advantage of the PDS process is that it provides an early opportunity to obtain our assessment of the suitability of a new device for the de novo process, our preliminary perspective on the likely classification, as well as feedback on the evidence, including performance and/or clinical data, that will likely be necessary to support the de novo petition. By obtaining this early feedback, you are more likely to optimize your resources in collecting the necessary safety and effectiveness evidence needed to support a de novo petition, without the need to perform additional tests. This should also facilitate the review of a subsequent de novo petition. We also believe that the PDS pathway will foster increased predictability and transparency in the de novo process and make the process more attractive to sponsors of de novo eligible devices.
Alternatively, you may choose to not submit a PDS. In this case, a de novo petition may be submitted within 30 days after a 510(k) for a device is found NSE. The success of a de novo petition that is filed without a prior PDS will depend on how adequately you identify the risks and special controls (if applicable) and how well you define and collect adequate data to provide reasonable assurance of safety and effectiveness. Note that a PDS should not be submitted once a 510(k) has been submitted, unless we specifically advise you to do so.
3.2.1 Pre De Novo Submission (PDS) -- 510(k)/De Novo Petition
Attachment 3 describes the format and content we suggest you use for a PDS. The PDS may be submitted early in the development process for a new device; however, we believe it is most useful after you have identified the proposed intended use and key aspects of the device design sufficient to permit a meaningful discussion on de novo suitability. In addition, the PDS should contain sufficient information to enable us to provide guidance on the test methods and protocols to be used for the collection of performance data.
After you submit your PDS, we may ask you for clarification or to provide more information, for example, to address protocol deficiencies or additional types of data that may be needed. We intend to request such information within 60 days of your initial submission of a PDS or your most recent response to such a request. Please note that our review of the PDS will generally not include the review of performance data. You may also request a meeting with us after a PDS has been submitted to discuss the content and review process for the submission. The requested meeting time should be no earlier than 30 days after your submission.
We intend to issue a suitability letter within 60 days after receipt of all information necessary to complete the review. The suitability letter will specify whether or not the device appears to be suitable for consideration via the de novo process and, if so, the likely class, likely special controls (if any), and necessary types of performance data.3 If the device does not appear to be suitable for the de novo process, the non-binding suitability letter will indicate the reasons for this assessment.
If at the end of the PDS review, we inform you that the new device appears to be suitable for the de novo process, the next step is for you to concurrently submit both a 510(k) and a de novo petition that should contain the information and data described in the PDS suitability letter. Once FDA sends this suitability letter, there is no time limit for sponsors to collect the information necessary for the concurrent 510(k)/de novo petition submission.
We recognize that some of the information submitted in a traditional or abbreviated 510(k)4 is not relevant for a de novo petition. As a result, we suggest that you identify these sections and respond by stating: “We are seeking concurrent FDA review of a de novo petition.”
The concurrent de novo petition should contain information consistent with Attachment 3; however, if the information has been submitted in the concurrent 510(k), it is appropriate for the petition to reference this information instead of repeating it within the petition.
The “PDS followed by 510(k)/de novo petition” pathway as described above is outlined in Attachment 1.
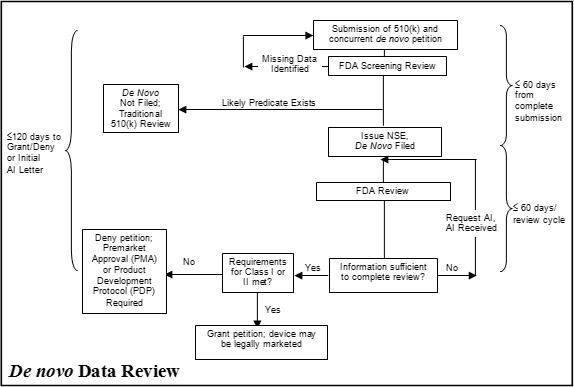
3.2.2 510(k) -- De Novo Petition
Alternatively, you may continue to use the existing de novo process, which begins with the submission of a 510(k) that results in an NSE determination. The de novo petition is required to be submitted within 30 days of the date-stamp issuance of the NSE letter. FDA will not review de novo petitions received more than 30 days after it issues the NSE letter. If you seek de novo review after this time period, you may submit a new 510(k) or a PDS for the device. The de novo petition should include or reference all information and evidence supporting the safety and effectiveness of the device, including the general and/or special controls which would provide reasonable assurance of safety and effectiveness. The de novo petition should establish the risk profile of the new device, the benefits of device use, and provide data demonstrating that general and/or special controls support a classification of Class I or Class II. See Attachment 3 for recommended content of a de novo petition.
The “510(k) followed by de novo” pathway is outlined in Attachment 2.
3.3 Address for Submission of the PDS and/or De Novo Petition
For devices regulated by CDRH, de novo petitions and PDSs should be submitted to:
U.S. Food and Drug Administration
Center for Devices and Radiological Health
Document Mail Center - WO66-G609
10903 New Hampshire Ave
Silver Spring, Maryland 20993-0002
For devices regulated by CBER, de novo petitions and PDSs should be submitted to:
U.S. Food and Drug Administration
Center for Biologics Evaluation and Research
Document Control Center (HFM-99)
1401 Rockville Pike, suite 200N
Rockville, Maryland 20852-1448
4. FDA Review Process for De Novo
4.1 Pre De Novo Submission (PDS)
Our preliminary review of the PDS is to assess whether or not the submission contains adequate information to permit review for de novo classification suitability (e.g., sufficient device description, likely special controls for Class II, etc.). If you fail to include adequate information to allow us to proceed with the review, we plan to ask for further information within 60 days of your submission or your complete response to a previous information request. If you do not respond to our information request within 180 days, we will consider your PDS submission to be withdrawn.
Once you have provided adequate information, we intend to perform a suitability review to analyze the existence of likely predicate devices and whether the new device appears to be within a type that FDA has either classified by regulation into class III based on risk, or approved in a premarket approval application (PMA). If a likely predicate device, class III classification regulation, or device type for which FDA has approved a PMA is identified, we will stop the PDS review and send a letter indicating that the new device does not appear to be suitable for de novo and the reason for this assessment5.
If we are unable to find a likely predicate device, class III classification, or approved PMA for the new device type, we intend to continue our review of the PDS to analyze whether general and/or special controls are likely able to provide reasonable assurance of safety and effectiveness. We may follow up with questions regarding any identified special controls and/or protocols for collection of safety and effectiveness data.
If it appears likely that general and/or special controls can provide reasonable assurance of safety and effectiveness, the resulting non-binding suitability letter will specify the likely class, likely special controls (if any), and necessary types of performance data. If we do not believe that general and/or special controls can provide reasonable assurance of safety and effectiveness, the PDS letter will indicate that the device does not appear to be suitable for de novo based on the information provided and specify the reasons for this assessment (e.g., it does not appear likely that there would be sufficient information to establish special controls to provide reasonable assurance of safety and effectiveness). After receiving a PDS letter indicating that the device does not appear suitable for the de novo process, you may choose to collect additional evidence to attempt to address our concerns and then submit a new PDS.
4.2 510(k)s Followed by De Novo Petitions
If, at the end of our review of a 510(k), we determine that the device is NSE due to a new intended use or new types of technology issues, we will consider whether the new device may be suitable for review under the de novo process. This review will occur per standard review practices for 510(k)s and in accordance with current performance goals. If the new device appears to present a low to moderate risk and there is sufficient information in the 510(k) to suggest that general and special controls may provide reasonable assurance of safety and effectiveness, we may indicate in the NSE letter that the product may be appropriate for the de novo process under section 513(f)(2) of the FD&C Act.
4.3 Post-PDS Concurrent 510(k)/De Novo Petition
As noted above, the 510(k) and de novo petition review is streamlined if you previously submitted a PDS and the new device appeared to be suitable for de novo. For 510(k)s submitted concurrently with a de novo petition following a PDS suitability review (see Attachment 1 and Section 3.2.1), we intend to first complete a screening review within 20 days of receipt of the concurrent submission to assure that you have submitted the necessary data. The screening review is only to determine completeness of the submission. If you have not submitted the information and/or data necessary to permit review , we will notify you as to which information and/or data are missing, and the review clock will reset to 60 days upon receipt of complete information. In the event you do not provide the requested information within 180 days, we will consider your concurrent 510(k) and de novo petition to be withdrawn.
Upon successful completion of the screening review, FDA will begin the in-depth review of the submission. First we will analyze whether a likely predicate device for the new device exists, for example, if one has been established through the de novo process in the interim since the PDS suitability review. If a likely predicate device exists, we plan to notify you that your de novo petition has not been filed and request information to enable us to review the 510(k) under the substantial equivalence standard. If no likely predicate device exists, we plan to issue an NSE determination for the 510(k) within 60 days of receipt of a complete concurrent submission. The NSE letter will indicate that we have filed your de novo petition and we will continue our review. See Section 4.4 for petition review process.
4.4 De Novo Petition
Under the existing process (no PDS), if you file a de novo petition within 30 days of the NSE letter, we conduct an initial review to confirm that the petition contains the necessary information (see Attachment 3 for recommended content). If the petition is missing significant information and/or data necessary to determine whether general and/or special controls can provide reasonable assurance of safety and effectiveness, we may immediately issue a denial of the petition. If you have submitted all information necessary to determine whether general and/or special controls can provide reasonable assurance of safety and effectiveness, we will continue the petition review.
Under either pathway (510(k) followed by de novo petition or PDS followed by concurrent 510(k)/de novo petition), if we identify issues requiring clarification after we have filed the petition, we may issue an additional information (AI) letter or request information via interactive review. Issuance of an AI letter resets the review clock, and once you provide the requested information, the review cycle is an additional 60 days.6 If you fail to provide a complete response to an AI request within 180 days, we will consider the de novo petition to be withdrawn. A petition withdrawn or denied due to failure to submit adequate information will require another 510(k) (and, optionally, a preceding PDS) to reinitiate review of the device under the de novo process.
If general and/or special controls are insufficient to provide reasonable assurance of safety and effectiveness or the information and/or data provided do not support safety and effectiveness for the intended use, we will issue a denial of the petition. In this case, the new device will remain in class III and require premarket approval under section 515 of the FD&C Act prior to marketing.
If general and/or special controls are adequate to provide reasonable assurance of safety and effectiveness, we will grant the de novo petition. If a de novo petition is granted, we will issue a written order granting the petition and specifying the classification of the new device into either class I or class II. Once you receive a written order granting the de novo petition, you may immediately begin marketing the device. We will follow the written order with a regulation and associated federal register notice announcing the device identification and classification. For class II devices, we will also identify special controls. When the special controls are straightforward or in cases where we do not anticipate that there will be many devices of the same type, we may identify the special controls in detail in the accompanying regulation. For more complex devices or in cases where we expect a significant number of devices of the same type, we will typically generate a special controls guidance document to accompany the regulation.
If a de novo petition is granted, we intend to make the written order granting the petition and a summary of our review of the petition available on the FDA website. The summary will contain a description of any special controls so that manufacturers who want to use the device as a predicate device for their new device have timely access to this information.
Attachment 1
Pre De Novo Submission (PDS) Followed by 510(k)/De Novo Petition Pathway

Attachment 2
510(k) Followed by De Novo Petition Pathway

Attachment 3
Recommended Content of a Pre de novo Submission (PDS) and a De novo Petition
The cover letter for a PDS or de novo petition should clearly identify “De Novo Petition” or “Pre De Novo Submission.”
If significant data for any of the sections below are contained in a related submission, you may provide cross-reference to the information. Any cross-references should include applicable volume/section/page numbers as appropriate.
A. INFORMATION SUBMITTED IN DE NOVO PETITION OR PDS
Administrative Information:
Applicant name, contact name, address, phone, fax, e-mail. For a PDS, if you are requesting a meeting with us to discuss the submission, indicate at least three (3) proposed dates/times, the type of meeting requested (in-person or teleconference), and the proposed agenda. Proposed meeting dates should be at least 30 days after the date of your PDS submission.
Regulatory History:
Describe any prior submissions to FDA for the new device, including any 510(k)s and related NSE decisions, IDEs, PDSs, and/or previously denied de novo petitions.
Device Information and Summary:
Device name, device description, indications for use statement (including prescription versus over the counter), and a description of all main functions, technological characteristics, components, and accessories. Include a summary of the directions for use/usage instructions. Identify the target population including demographics information, diseases, and/or symptoms to be treated, etc.
Classification Recommendation:
Recommended Class [I or II] and recommended applicability of 510(k) requirement [exempt or not exempt]. Describe why you believe general and/or special controls are adequate to provide reasonable assurance of safety and effectiveness. If you believe you should be exempt from 510(k), justify why premarket notification should not be required.
Supporting Protocols and/or Data:
A PDS should be accompanied by all protocols for bench and clinical testing, including targeted performance levels and anticipated risks, that will demonstrate that general and/or special controls are sufficient to provide reasonable assurance of safety and effectiveness. If test results are available at the time of a PDS submission, you are encouraged to submit this information as well. For a de novo petition, all final protocols and supporting data should be submitted.
Summary of Benefits:
Provide information supporting the effectiveness of the new device. Cite the available data/studies supporting effectiveness. For a PDS, briefly describe any ongoing and/or planned protocols/studies that need to be completed to collect the necessary data to establish effectiveness. For a de novo petition, summarize the studies completed and how they support effectiveness.
Summary of Known and Potential Risks to Health:
List each risk and identify the reason for each risk (tracing back to risk analysis, clinical testing, etc.). For a PDS, briefly describe any ongoing and/or planned protocols/studies that need to be completed to collect the necessary data to establish the device's risk profile. For a de novo petition, summarize the studies completed and how they support safety.
Risk and Mitigation Information:
Provide a table showing the proposed mitigation(s) for each risk (for a PDS, base this table on the best available information at the time of the submission). Highlight which mitigations are general controls or special controls. Provide details on each recommended special control (e.g., specific testing required, etc.) in the submission.
| Identified Risk | Recommended Mitigation Measures | Control Type (General or Special) |
|---|---|---|
| EXAMPLE: Adverse tissue reaction | Biocompatibility | Special |
| EXAMPLE: Device failure | Nonclinical Analysis and Testing | Special |
| Labeling | Special | |
| Report to FDA | General | |
| EXAMPLE: Failure to properly interpret test results | Labeling | Special |
If special controls are required, we encourage you to submit proposed special controls language as it will appear in the accompanying regulation or a draft special controls guidance document as part of the de novo petition.
Device Labeling:
If draft labeling7 is available at the time of the PDS submission, we encourage you to provide the labeling as part of the PDS. You should include proposed device labeling in a de novo petition.
B. ADDITIONAL INFORMATION TO BE SUBMITTED IN A PRE-510(k) PDS
For a PDS that is submitted before a device has been reviewed under the 510(k) process, provide the following additional section:
Classification Summary:
Describe why the new device does not have a predicate device, fit into an existing classification regulation, or be of a type that has been approved in a PMA. For potential predicate devices with similar technology and/or indications, include discussion of why the new device is not substantially equivalent.
C. ADDITIONAL INFORMATION TO BE SUBMITTED IN A POST-PDS De Novo Petition
For a de novo petition that is submitted concurrently with a 510(k) after a determination of de novo suitability under a PDS, provide the following additional section:
Change Summary:
Describe all changes since the PDS submission. This summary should include changes to the device as well as changes to test protocols and/or labeling.
1 The process has been termed “de novo” because it requires the agency to evaluate novel devices anew, from an independent, risk-based standpoint under the criteria at section 513(a)(1) of the FD&C Act, after FDA has made an NSE determination under the comparative 510(k) substantial equivalence standard.
2 To be consistent with other guidance documents relating to the 510(k) process, this guidance uses the phrase "new device" to refer to the device for which marketing authorization is sought, i.e., the device that is the subject of de novo classification review. This phrase is not intended to imply that there is an “old” or predicate device to which a comparison may be made under section 510(k). This phrase should also not be confused with use of the term “new” or “novel” to refer to types of devices that may be reviewed through de novo classification.
3 See Section 4.3 for a discussion of this suitability determination. Any special controls identified during the PDS process are contingent on collection of adequate performance data. If, for example, new safety or effectiveness issues arise during subsequent clinical testing of the device, additional special controls may be identified or the device may no longer be suitable for classification under the de novo process.
4 See Guidance for Industry and FDA Staff: Format for Traditional and Abbreviated 510(k)s. Specifically, we refer to Sections 4, 5, 7 and 12.
5 We note that, with respect to a class III device type, you may submit a reclassification petition under section 513(f)(3) of the Act, 21 U.S.C. 360c(f)(3), requesting the issuance of an order classifying the device in class I or class II. The content of and process for reclassification petitions is described in 21 CFR 860, Subpart C.
6 In rare instances, we may seek input on a de novo petition from a Classification Panel of the FDA Medical Devices Advisory Committee. In such instances, we will likely need to extend overall review timelines for de novo petitions.
7 Labeling is defined in section 201(m) of the FD&C Act, 21 U.S.C. 321(m), as “all labels and other written , printed or graphic matter (1) upon any article or any of its containers or wrappers, or (2) accompanying such article.” Labeling may include package inserts, instructions for use (for patient and/or physician, as applicable), service manuals (if required), etc.
