


Food
WITHDRAWN Significant Scientific Agreement in the Review of Health Claims for Conventional Foods and Dietary Supplements
This guidance no longer applies. It was withdrawn on January 16, 2009. For the Federal Register Notice of the withdrawal, see
http://www.fda.gov/Food/LabelingNutrition/FoodLabelingGuidanceRegulatoryInformation/
RegulationsFederalRegisterDocuments/ucm114304.htm
2001 (Rev. 2)
December 22, 1999
Guidance for Industry
Significant Scientific Agreement in the Review of Health Claims for Conventional Foods and Dietary Supplements
Comments and suggestions regarding this document should be submitted by February 22, 2000, to Dockets Management Branch (HFA-305), Food and Drug Administration, 5630 Fishers Lane, rm 1061, Rockville, MD 20852. All comments should be identified with the docket number 99D-5424.
For questions concerning the content of the document contact Sharon Ross at 202-205-4168.
Additional copies of this guidance document are available upon written request from the Office of Special Nutritionals (HFS-450), Food and Drug Administration, 200 C Street SW, Washington, DC 20204, by calling 202-205-4168, by faxing a request to 202-205-5295, or from the Internet at http://www.cfsan.fda.gov/~dms/guidance.html#lab.
U. S. Department of Health and Human Services
Food and Drug Administration
Center for Food Safety and Applied Nutrition
December 22, 1999
Table of Contents
I. BACKGROUND INFORMATION
II. SCIENTIFIC REVIEW OF HEALTH CLAIMS
B. Performing Reliable Measurements
C. Evaluating Individual Studies
D. Evaluating the Totality of the Evidence
E. Assessing Significant Scientific Agreement
Figure 1. Type of Study Evaluated During Health Claim Review
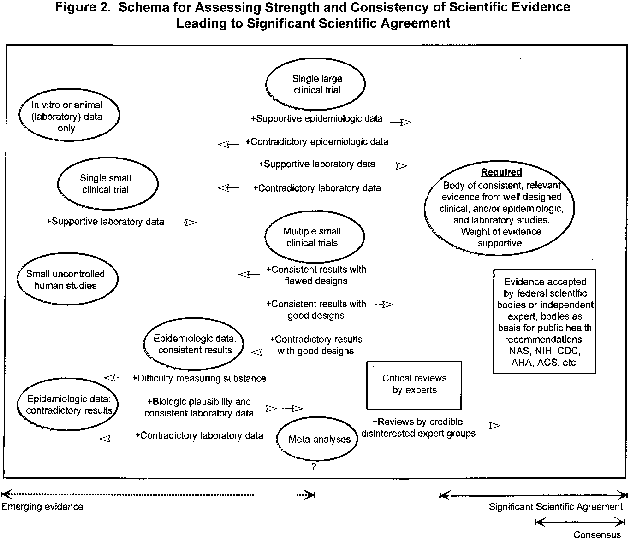
Figure 2. Schema for Assessing Strength and Consistency of Significant Scientific Agreement
Introductory Note
This guidance has been prepared by the Office of Special Nutritionals in the Center for Food Safety and Applied Nutrition at the Food and Drug Administration (FDA), based on the report of the FDA Food Advisory Committee (FAC) Working Group on Significant Scientific Agreement. This guidance represents the agency's current thinking on the meaning of the significant scientific agreement standard in section 403(r)(3) of the Federal Food, Drug, and Cosmetic Act (21 U.S.C. § 343(r)(3)) and 21 CFR § 101.14(c). It is being issued as level 1 guidance for immediate implementation in accordance with FDA's good guidance practices (62 FR 8961, February 27, 1997). The guidance document does not create or confer any rights for or on any person and does not operate to bind FDA or the public. An alternative approach may be used if such approach satisfies the requirements of the applicable statute, regulations or both.
This guidance document addresses the significant scientific agreement standard, which FDA uses to evaluate the scientific evidence supporting health claim petitions about the relationship between a food substance and a disease or health-related condition. The guidance document describes the scientific review approach FDA has taken in previous health claim reviews and incorporates the recommendations of the FDA FAC Working Group on Significant Scientific Agreement. This approach is used by FDA scientists in their review of health claims and should be considered as guidance by those compiling health claim petitions. The scientific principles described in this document should also be useful to those designing studies to support health claim petitions.
FDA's determination on significant scientific agreement represents the agency's best judgment as to whether qualified experts would likely agree that the scientific evidence supports the substance/disease relationship that is the subject of a proposed health claim. The significant scientific agreement standard is intended to be a strong standard that provides a high level of confidence in the validity of a substance/disease relationship. Significant scientific agreement means that the validity of the relationship is not likely to be reversed by new and evolving science, although the exact nature of the relationship may need to be refined. Application of the significant scientific agreement standard is intended to be objective, in relying upon a body of sound and relevant scientific data; flexible, in recognizing the variability in the amount and type of data needed to support the validity of different substance/disease relationships; and responsive, in recognizing the need to re-evaluate data over time as research questions and experimental approaches are refined. Significant scientific agreement does not require a consensus or agreement based on unanimous and incontrovertible scientific opinion. However, on the continuum of scientific discovery that extends from emerging evidence to consensus, it represents an area on the continuum that lies closer to the latter than to the former.
Before significant scientific agreement can be assessed, a number of sequential threshold questions are addressed in the review of the scientific evidence:
- Have studies appropriately specified and measured the substance that is the subject of the claim?
- Have studies appropriately specified and measured the disease that is the subject of the claim?
- Are any and all conclusions about the substance/disease relationship based on the totality of publicly available scientific evidence?
The assessment of significant scientific agreement then derives from the conclusion that there is a sufficient body of sound, relevant scientific evidence that shows consistency across different studies and among different researchers and permits the key determination of whether a change in the dietary intake of the substance will result in a change in a disease endpoint.
The specific topics addressed in this guidance document are: identifying data for review, performing reliable measurements, evaluating individual studies, evaluating the totality of the evidence, and assessing significant scientific agreement. Other aspects of and requirements for the health claim authorization process are described in the Code of Federal Regulations, in 21 CFR § 101.14 and 21 CFR § 101.70.
Major considerations in the scientific review process for health claims are highlighted in bold-face type. For each step in the process, details of the issues that should be considered are provided. Explanatory comment, illustrative discussion points, and examples of application of criteria or requirements, as demonstrated by past health claim authorization reviews, are provided in italics.
Background Information
The Nutrition Labeling and Education Act of 1990 (NLEA) was designed to give consumers more scientifically valid information about the foods they eat (1). Among other provisions, NLEA authorized FDA to allow statements that describe the relationship between a nutrient and a disease or health-related condition to appear in the labeling of foods, including dietary supplements. Such statements about substance/disease relationships are known as "health claims." FDA has defined the term "substance" by regulation as a specific food or component of food. An authorized health claim may be used on both conventional foods and dietary supplements, assuming that the substance in the product and the product itself meet the appropriate standards. Health claims are directed to the general population or designated subgroups (e.g., the elderly) and are intended to assist the consumer in maintaining healthful dietary practices.
When FDA decides whether to authorize a health claim, it evaluates, among other considerations, whether the evidence supporting the relationship that is the subject of the claim meets the significant scientific agreement standard. This standard derives from 21 U.S.C. § 343(r)(3)(B)(i), which provides that FDA shall authorize a health claim to be used on conventional foods if the agency "determines, based on the totality of the publicly available scientific evidence (including evidence from well-designed studies conducted in a manner which is consistent with generally recognized scientific procedures and principles), that there is significant scientific agreement, among experts qualified by scientific training and experience to evaluate such claims, that the claim is supported by such evidence." This scientific standard applies to conventional food health claims by statute; FDA applied the same standard to dietary supplement health claims by regulation. See 21 CFR § 101.14(c).
The NLEA identified 10 substance/disease relationships for initial consideration(1). Of these, significant scientific agreement was determined to exist for eight of the relationships, and health claims describing these relationships on food labels were authorized in 1993. The legislation also permits any interested person to petition FDA to issue a regulation regarding a health claim. Additional health claims have been authorized in response to such petitions.(1)
Since NLEA was enacted, several groups have evaluated the health claim authorization process, including the interpretation of significant scientific agreement. One of these evaluations was a 2-year Keystone Center dialogue among representatives from academia, industry, consumer groups, and government. The dialogue and resulting report affirmed the principles and approach FDA had been using to authorize health claims(2). The Commission on Dietary Supplement Labels examined the health claim authorization process for dietary supplements and also generally expressed agreement with FDA's approach in its report (3). Following the Keystone dialogue, the FDA FAC convened a number of working groups in 1996 to address issues raised and recommendations made during the dialogue. The FAC Working Group on Significant Scientific Agreement was charged with developing a guide for preparing health claim petitions. In response to the recent decision of the United States Court of Appeals for the District of Columbia Circuit in Pearson v. Shalala, 164 F.3d 650 (D.C. Cir. 1999), which required FDA to clarify the meaning of significant scientific agreement, the focus of the FAC Working Group shifted to the scientific review of data for health claims and the interpretation of the significant scientific agreement standard. The final report of the FAC Working Group on Significant Scientific Agreement, entitled "Interpretation of Significant Scientific Agreement in the Review of Health Claims," was made public during the FAC meeting of June 24-25, 1999. (See http://vm.cfsan.fda.gov/~dms/facssa.html for a copy of the Working Group's report.) Following additional comment by the FAC, FDA adopted the recommendations proposed by the Working Group on Significant Scientific Agreement. This guidance document is based on the FAC Working Group report but has been expanded and edited to clarify and more fully explain some topics. The guidance represents the agency's current thinking on the meaning of significant scientific agreement in 21 U.S.C. § 343(r)(3)(B)(i) and 21 CFR § 101.14(c).
Footnote:
- In 1997, Congress enacted the Food and Drug Administration Modernization Act, which established an alternative authorization procedure for health claims based on authoritative statements from certain federal scientific bodies or from the National Academy of Sciences. As of December 1999, one health claim had been authorized under this alternative procedure. This guidance document does not address that alternative procedure.
Scientific Review of Health Claims
The scientific review process FDA uses to evaluate health claims is comprehensive and focuses first on review of individual studies. After identifying relevant, good quality studies and assessing their strengths and weaknesses, the agency conducts a more comprehensive review based on the body of evidence as a whole. Considerations in the scientific review of health claims are detailed below.
The standard of scientific validity for a health claim includes two components: 1) that the totality of the publicly available evidence supports the substance/disease relationship that is the subject of the claim, and 2) that there is significant scientific agreement among qualified experts that the relationship is valid.
FDA's evaluation of the evidence supporting a health claim is based on the totality of publicly available data. Because of the limitations of the various research methods that can be used to study substance/disease relationships, it is not possible to specify the type or number of studies needed to support a health claim. In addition, each relationship involves a unique set of confounders (see discussion below) and measurement issues.
In addition to limitations imposed by available research methods, another limitation frequently encountered is the dependence on publicly available data derived from studies that were not specifically designed or conducted for the purpose of supporting a health claim. Thus, in the agency's review of health claims, the usefulness, relevance, and generalizability of such studies to the health claim under consideration are carefully evaluated, especially in terms of specification and measurement of the substance and disease whose relationship is the subject of the claim.
A. Identifying Data for Review
The first step in preparing or reviewing a health claim petition is to identify all relevant studies.
The types of studies considered in a health claim review include human studies and frequently also include "pre-clinical" evidence, e.g., in vitro laboratory investigations and other mechanistic studies. Studies of humans can be divided into two types: interventional studies and observational studies.
In an interventional study, the investigator controls whether the subjects receive an exposure or an intervention whereas in an observational study, the investigator does not have control over the exposure or the intervention. In general, interventional studies provide the strongest evidence for an effect.
Regardless of the inherent strengths and weaknesses of a study design, the overall quality and relevance of each individual study is paramount in assessing its contribution to the weight of the evidence for the proposed substance/disease relationship.
- Interventional studies
The "gold standard" of interventional studies is the randomized controlled clinical trial.
In a randomized controlled trial, subjects similar to each other are randomly assigned either to receive the intervention or not to receive the intervention. As a result, subjects who are most likely to have a favorable outcome independent of any intervention are not preferentially selected to receive the intervention being studied (selection bias). Bias may be further reduced if the researcher who assesses the outcome does not know which subjects received the intervention (blinding). Randomized controlled clinical trials are not an absolute requirement to demonstrate significant scientific agreement in all cases, but are considered the most persuasive and given the most weight. A single large, well-conducted and controlled clinical trial could provide sufficient evidence to establish a substance/disease relationship, provided that there is a supporting body of evidence from observational or mechanistic studies.
Interventional studies for foods may differ from those for drugs. Unlike drug studies, food interventional trials may have additional confounders secondary to using a food substance as the intervention (see discussion below). In addition, it may not be possible to use a placebo control group for food studies, and subjects in such studies may not be blinded to the intervention. As a result of the greater likelihood for confounders and bias, interventional studies with foods may generate data that have less certainty than data from drug interventional studies.
- Observational studies
- Cohort studies compare the outcome of subjects who have received a specific exposure with the outcome of subjects who have not received that exposure.
- In case-control studies, subjects with the disease are compared to subjects who do not have the disease (control group). Subjects are enrolled based on their outcome rather than based on their exposure.
- In cross-sectional studies, at a single point in time the number of individuals with a disease who have received a specific exposure is compared to the number of individuals without the disease who did not receive the exposure.
- Uncontrolled case series studies depict outcomes in a group without comparing to a control group.
- Time-series studies compare outcomes during different time periods, e.g., whether the rate of occurrence of a particular outcome during one five-year period changed during a subsequent five-year period.
- In ecological studies, the rate of a disease is compared across different populations. Investigators seek to identify population traits that may cause the disease.
- Descriptive epidemiology refers to study designs that assess parameters related to the frequency and distribution of disease in a population, such as the leading cause of death.
- Case reports describe observations of a single subject or a small number of subjects.
A common weakness of observational studies is the limited ability to ascertain the actual food or nutrient intake for the population studied. Observational data are also generally restricted to identifying associations between food substances and health outcomes, and often do not provide a sufficient basis for determining whether a substance/disease association reflects a causal rather than a coincidental relationship.
- Research synthesis studies
- Animal and in vitro studies
B. Performing Reliable Measurements
Appropriate measurement, of both the substance and the disease or health-related condition, is a key factor in the review of data for health claims.
Assessing the effects of diet on human health is limited by a variety of measurement issues: the use of biomarkers, the difficulty of identifying and measuring the food substance that provides the effect, the difficulty of accurately measuring dietary intake, and the difficulty of distinguishing the effects of diet on a disease from those of other variables, such as weight change, physical activity, or environmental factors.
- Biomarkers
- Identifying and measuring the food substance
Studies that examine dietary components often focus on the intake of the substance of interest as part of a food or a total diet, or may infer intake as part of post-hoc evaluations of the data. Therefore, isolating the effect of the substance can be a critical consideration in authorizing a health claim. Common difficulties involve separating the effect of the food substance from the food itself, or the use of measures that reflect heterogeneous or poorly defined food substances. Without evidence that the substance, rather than the overall diet or specific foods in the diet, is responsible for the benefit, the linkage between the substance and the disease cannot be established.
FDA applied this principle during evaluations of the initial 10 substance/disease relationships in 1990-1992. In the case of claims related to omega-3 fatty acids, fiber, and antioxidant vitamins, there was considerable measurement overlap between the food containing the substance and the substance itself, or there were concomitant changes in other dietary components. Fiber was poorly defined and/or a heterogeneous mixture as measured in research available at the time of the initial health claim review. For example, as noted during the health claim review for fiber and heart disease, the objective of the protocols of many studies was to evaluate the effectiveness of relatively large amounts of a single type of food or fiber source rich in soluble fiber (e.g., baked beans), rather than to examine total soluble dietary fiber intakes or to specifically identify the chemical and physical characteristics of soluble fiber that are most effective in lowering blood cholesterol levels. Thus, the effects could not be attributed to the fiber. Moreover, in some studies large amounts of foods (e.g., 1-2 cups of baked beans) were added to diets; these dietary changes were often accompanied by lower calorie intakes with resultant weight loss, which has an independent impact on the risk of developing heart disease.
- Assessment of dietary intake
- Distinguishing the effects of diet from other variables
C. Evaluating Individual Studies
The evaluation of study design, protocol, measurement, and statistical issues for individual studies serves as the starting point from which FDA determines the overall strengths and weaknesses of the data and assesses the weight of the evidence.
FDA's review of individual studies on substance/disease relationships generally follows the approaches outlined in the Guide to Clinical Preventive Services (9) and Diet and Health (8).
The persuasiveness of a study depends on the quality of the study.
- Bias and confounders
- Quality assessment criteria
- Adequacy and clarity of the design
Were the questions to be answered by the study clearly described at the outset?
Was the methodology used in the study clearly described and appropriate for answering the questions posed by the study?
Was the duration of the study intervention or follow-up period sufficient to detect an effect on the outcome of interest?
Were potential confounding factors identified, assessed, and/or controlled?
Was subject attrition (subjects leaving the study before the study is completed) assessed, explained, and reasonable?
- Population studied
Was the sample size large enough to provide sufficient statistical power to detect a significant effect? (If the study is underpowered, it may be impossible to conclude that the absence of an effect is not due to chance.)
Was the study population representative (for factors such as age, gender distribution, race, socioeconomic status, geographic location, family history, health status, and motivation) of the population to which the health claim will be targeted?
Were criteria for inclusion and exclusion of study subjects clearly stated and appropriate?
Were recruitment procedures that minimized selection bias used?
- Assessment of intervention or exposure and outcomes
Were analytical methodology and quality control procedures to assess dietary intake adequate?
Was the dietary intervention or exposure well defined and appropriately measured? (See discussion above.)
For intervention studies, was an appropriate level of intake (i.e., the level hypothesized to be effective) for the food substance of interest planned, monitored, and achieved?
Were the background diets to which the test substance was added, or the control and interventional diets, adequately described, measured, and suitable?
Was a "lead-in" period employed for dietary interventions? (Because changes in the diet may induce compensatory metabolic changes, the effect of an intervention should be measured after stabilization has occurred, i.e., a lead-in period.)
In studies with cross-over designs, was there an appropriate "wash-out" period (period during which subjects do not receive an intervention) between dietary treatments? (Lack of a sufficient wash-out period between interventions may lead to confusion as to which intervention produced the health outcome.)
Were the form and setting of the intervention representative of the "real world?"
Were other possible concurrent changes in diet or health-related behavior (weight loss, exercise, alcohol intake, smoking cessation) during the study that could account for the outcome identified, assessed, and/or controlled?
Were the disease outcomes well defined and appropriately measured? If biomarkers (intermediate or surrogate endpoint markers) were measured, has their relevance to disease outcomes been validated?
Were efforts made to detect harmful as well as beneficial effects? (For example, increasing the consumption of some food substances may increase the risk of a chronic disease, and extracting or concentrating some food substances may render them injurious to health.)
- Statistical methods
-
Were appropriate statistical analyses applied to the data?
Was "statistical significance" interpreted appropriately? (For example, differences that are not statistically significant should be described as not demonstrating a difference rather than as showing a trend.)
Were relative and absolute effects distinguished?
- Summary of the evidence
D. Evaluating the Totality of the Evidence
Evaluating the totality of the evidence means evaluating whether it permits the key determination of whether a change in the dietary intake of the substance will result in a change in a disease endpoint.
After identifying relevant, good quality studies and assessing and summarizing their strengths and weaknesses, FDA conducts a more comprehensive review based on the body of evidence as a whole. Petitioners should be sure that the conclusions the petition draws regarding the association between nutritional exposures or interventions and outcomes are objectively based on the totality of the evidence, and that interpretations are limited to the research conducted, without inappropriate extrapolations beyond the available evidence.
- Determining the strength of the substance/disease association
- The first consideration in judging the body of evidence is determining whether most of the evidence is derived from more persuasive classes of study designs.
- Another contribution to the strength of the evidence is the number of studies in support of the association.
- Consistency of results across different settings and types of populations also bolsters the strength of an association.
- Finally, if the magnitude of the effect is large, yielding strong statistical significance and narrow confidence intervals, evidence of an association is bolstered and the association is more likely to have clinical significance.
- Determining the strength of the substance/disease relationship (inferring that a causal relationship exists)
- Strength of association is sometimes described as relative risk. Relative risk is the ratio between the rate of disease for subjects exposed to the substance and the rate for subjects not exposed. The larger the relative risk, the more likely that ingesting the substance is causally related to the health outcome.
- Consistency of association means that the same association is found across several studies and among various population groups.
- Independence of association refers to the extent to which the association relates to the exposure or intervention being studied versus the extent to which the association relates to a variable other than the exposure or intervention.
- Dose-response relationship means that greater effects occur with greater exposures to the substance.
- Temporal relationship means that the exposure consistently precedes the outcome.
- Effect of dechallenge means that subjects from whom the intervention has been withdrawn demonstrate a reversal of the associated outcome.
- Specificity means the degree to which the substance is associated only with the disease in question. The more specific an association, the more likely the association is causal. However, lack of specificity may not be a critical factor in the assessment of substance/disease relationships because many etiological agents cause more than one disease, and many diseases have multifactorial causes.
- Biological plausibility means that there is a biological explanation for the causal relationship. Although biological plausibility is not necessary to infer causality, it enhances the case.
- Determining the weight of the evidence as a whole
- Does the evidence in support of the substance/disease relationship outweigh the evidence against it? In considering this question, appropriate weight should be given to studies that are more persuasive because of the quality of the study design, conduct, and analysis.
- Is the available body of evidence sufficient to permit the conclusion that a change in the dietary intake of the substance will result in a change in the disease endpoint?
E. Assessing Significant Scientific Agreement
Assessing significant scientific agreement relies on judging the extent of agreement among qualified experts.
Significant scientific agreement refers to the extent of agreement among qualified experts in the field. In the process of scientific discovery, significant scientific agreement occurs well after the stage of emerging science, where data and information permit an inference, but before the point of unanimous agreement within the relevant scientific community that the inference is valid. The significant scientific agreement standard is met when the validity of the relationship is not likely to be reversed by new and evolving science, although the exact nature of the relationship may need to be refined over time. Significant scientific agreement can be achieved when the validity of a substance/disease relationship is supported by the conclusions of federal government scientific bodies; conclusions of independent, expert bodies may also be relevant. When such conclusions are not available (for instance, if the data supporting a proposed health claim are relatively new and have not yet been reviewed by an independent, expert panel or body), a compelling and relevant body of evidence may nonetheless cause the agency to conclude that significant scientific agreement exists.
- Significant scientific agreement depends on the strength and consistency of the evidence.
Figure 2 provides a graphical representation of the interplay of considerations that contribute to determining whether the significant scientific agreement standard for a substance/disease relationship has been met. It illustrates the manner in which evaluations of the various types and amounts of data that may exist for a substance/disease relationship are combined to assess the overall strength and consistency of the scientific evidence. The schema also demonstrates that the significant scientific agreement standard is one that is objective, flexible, and responsive by illustrating the variety of combinations of data from different types of good quality studies that may give rise to a body of evidence sufficient to establish the validity of a substance/disease relationship.
In determining whether there is significant scientific agreement, FDA takes into account the viewpoints of qualified experts outside the agency, if evaluations by such experts have been conducted and are publicly available. For example, FDA will take into account:
- review publications that critically summarize data and information in the secondary scientific literature;
- documentation of the opinion of an "expert panel" that is specifically convened for this purpose by a credible, independent body;
- the opinion or recommendation of a federal government scientific body such as the National Institutes of Health (NIH) or the Centers for Disease Control and Prevention (CDC); or the National Academy of Sciences (NAS); or an independent, expert body such as the Committee on Nutrition of the American Academy of Pediatrics (AAP), the American Heart Association (AHA), American Cancer Society (ACS), or task forces or other groups assembled by the National Institutes of Health (NIH).
References
1. Public Law 101-553, 104 Stat. 2353 codified at 21 USC § 343 (1994). Nutrition Labeling and Education Act. November 8, 1990.
2. The Keystone National Policy Dialogue on Food, Nutrition, and Health: Final Report. Keystone, CO: Keystone Press, 1996.
3. Commission on Dietary Supplement Labels. Commission of Dietary Supplement Labels Report to the President, Congress, and the Secretary of the Department of Health and Human Services. Washington, DC: Office of Disease Prevention and Health Promotion, DHHS, 1997.
4. Sacks HS, Berrier J, Reitman D, Ancona-Berk VA, Chalmers TC. Meta-analyses of randomized controlled trials. N Engl J Med 1987;316:450-455.
5. Sacks HS, Berrier J, Reitman D, Pagano D, Chalmers TC. Meta-analysis of randomized controlled trials: an update. In: Balder WC, Mosteller F, eds. Medical Uses of Statistics, 2nd ed. pp 427-442. Boston, MA: NEJM Books, 1992.
6. Sacks HS. Meta-analyses of clinical trials. In: Perman JA, Rey J, eds. Clinical Trials in Infant Nutrition, Nestle Nutrition Workshop Series, Vol 40, pp 85-99. Philadelphia, PA: Vevey/Lippincott-Raven Publishers, 1998.
7. Hasselblad V, Mosteller F, Littenberg B, Chalmers TC, Hunick MG, Turner JA, et al. A survey of current problems in meta-analysis. Discussion from the Agency for Health Care Policy and Research Inter-PORT Work Group on Literature Review/Meta-Analysis. Med Care 1995;33:202-220.
8. National Research Council. Diet and Health: Implications for Reducing Chronic Disease Risk. Washington, DC: National Academy Press, 1989.
9. Department of Health and Human Services, Office of Disease Prevention and Health Promotion. Report of the US Preventive Services Task Force: Guide to Clinical Preventive Services. 2nd ed. Washington, DC: Office of Public Health and Science, April, 1989.
10. Ahrens EH, Connor WE, eds. Symposium: Report of the Task Force on the Evidence Relating Six Dietary Factors to the Nation's Health. Am J Clin Nutr 1979;23(suppl):2621-2748.
11. Mohar D, Jadad AR, Tugwell P. Assessing the quality of randomized controlled trials: current issues and future directions. Int J Technol Assess Health Care 1996;12:125-208.
12. Albanes D, Heinonen OP, Huttunen JK, Taylor PR, Virtamo J, Edwards BK, Haapakoski J, Rautalahti M, Hartman AM, Palmgren J, et al. Effects of alpha-tocopherol and beta-carotene supplements on cancer incidence in the Alpha-Tocopherol Beta-Carotene Cancer Prevention Study. Am J Clin Nutr 1995;62(6 Suppl):1427S-1430S.

This document was issued on December 22, 1999.
For more recent information see Dietary Supplements
