
Información general sobre la leucemia linfoblástica aguda en adultos (LLA)
Incidencia y mortalidad
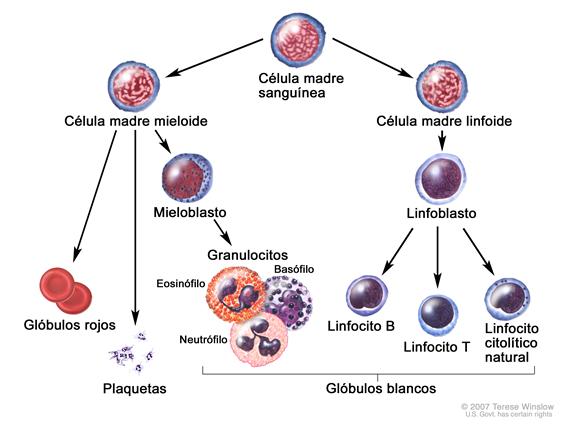
Anatomía
Genética molecular
Diagnóstico
Pronóstico y supervivencia
Efectos tardíos del tratamiento de la LLA en adultos
Ensayos clínicos en curso
La LLA (también llamada leucemia linfocítica aguda) es un tipo de leucemia muy fuerte que se caracteriza por la presencia de demasiados linfoblastos o linfocitos en la médula ósea y la sangre periférica. Se puede diseminar a los ganglios linfáticos, bazo, hígado, sistema nervioso central (SNC) y otros órganos. Sin tratamiento, la LLA avanza rápidamente.
Entre los signos y síntomas de LLA se encuentran:
- Debilidad o cansancio crónico.
- Fiebre o sudoración nocturna.
- Moretones o sangrar con facilidad (es decir, sangrado de las encías, parches morados en la piel o petequia [puntos planos bajo la piel]).
- Disnea.
- Pérdida de peso inesperada o anorexia.
- Dolor en los huesos o articulaciones.
- Ganglios linfáticos inflamados, especialmente en el cuello, axilas, o ingle, por lo general no dolorosos.
- Inflamación o incomodidad en el abdomen.
- Infecciones frecuentes.
La LLA se presenta tanto en niños como en adultos. Es el tipo de cáncer más común entre niños y el tratamiento resulta en una buena oportunidad de curación. Para los adultos, el pronóstico no es tan optimista. Este sumario se refiere a la LLA en adultos. (Para mayor información sobre la LLA en niños, consultar el sumario del PDQ Leucemia linfoblástica aguda infantil)
Incidencia y mortalidadCálculo del número de casos nuevos y muertes por LLA en los Estados Unidos en 2012:[1]
- Casos nuevos: 6.050.
- Muertes: 1.440.
Se presume que la LLA surge de la transformación maligna de las células progenitoras B o T.[2] Se ve con mayor frecuencia en los niños pero se puede presentar en cualquier edad. La enfermedad se caracteriza por la acumulación de linfoblastos en la médula o en varios sitios extramedulares, con frecuencia acompañado de supresión de la hematopoyesis normal. Las células leucémicas linfoblásticas B y T expresan antígenos de superficie muy similares a la evolución de sus respectivos linajes. Típicamente, las LLA de células precursoras B expresan CD10, CD19 y CD34 en su superficie junto con la dioxinucleotidiltransferasa terminal (TdT) nuclear, mientras que la LLA de células precursoras T por lo general expresan CD2, CD3, CD7, CD34 y TdT.
Por muchos años se ha reconocido que algunos pacientes que presentan leucemia aguda pueden tener una anomalía citogenética que no se puede distinguir citogenéticamente del cromosoma Filadelfia (Ph1).[3] El Ph1 se presenta solamente en 1 a 2% de los pacientes con leucemia mieloide aguda (LMA) pero se presenta en cerca de 20% de los adultos y en un porcentaje pequeño de niños con LLA.[4] En la mayoría de los niños y en más de la mitad de los adultos con LLA positivos al Ph1, la anomalía molecular es diferente de la que se presenta en la leucemia mielógena crónica (LMC) positiva al Ph1.
Muchos pacientes que presentan constancia molecular del gen de fusión bcr-abl, el cual caracteriza el Ph1, no exhiben un cromosoma anormal en el análisis citogenético. El bcr-abl se puede detectar sólo por hibridación fluorescente in situ (HFIS) o reacción en cadena de la polimerasa por transcriptasa inversa (RCP-TI) debido a que muchos pacientes tienen una proteína de fusión diferente de la que se encuentra en la LMC (p190 contra p210). Estos exámenes deberán llevarse a cabo cuando sea posible en pacientes con LLA, especialmente en aquellos con enfermedad de linaje de células B.
La LLA L3 está relacionada con una variedad de desplazamientos los cuales comprometen desplazamientos del protooncogén c-myc al locus del gen de inmunoglobulinas (t[2;8], t[8;12] y t[8;22]).
DiagnósticoLos pacientes con LLA podrían presentar una variedad de trastornos hematológicos que oscilan de pancitopenia a hiperleucocitosis. Además de los antecedentes y exámenes físicos los análisis iniciales deben incluir:
- Recuento sanguíneo completo con diferencial.
- Panel químico (que incluye ácido úrico, creatinina, nitrógenos sanguíneo y úrico, potasio, fosfato, calcio, bilirrubina y transaminasa hepática).
- Fibrinógenos y pruebas de coagulación como tamiz para la coagulación intravascular diseminada.
- Exámenes de detección cuidadosos en busca de pruebas de infección activa.
Con frecuencia se lleva a cabo de forma rutinaria una biopsia de médula ósea y aspirados aún en las LLA de células T para determinar la extensión del compromiso medular. Las células malignas se deben enviar a un estudio citogenético convencional, ya que la detección del reordenamiento Ph1 t(9;22), myc (en la leucemia de Burkitt y el reordenamiento MLL), añaden información pronóstica importante. La citometría de flujo se debe llevar a cabo para caracterizar los antígenos que definen la expresión de linaje y permitir la determinación de los subtipos específicos de la LLA. Además, para la enfermedad de células B, las células malignas deben analizarse utilizando PCR-RT e HFIS como prueba de del gen de fusión bcr-abl. Este último punto es de suma importancia, ya que un diagnóstico a tiempo de la LLA Ph1 cambaría de manera significativa el enfoque terapéutico.
No es rara la confusión del diagnóstico con el de la leucemia mielocítica aguda (LMA), leucemia de células pilosas y el linfoma maligno. Es de suma importancia efectuar un diagnóstico adecuado debido a las diferencias en el pronóstico y tratamiento entre la LLA y la LMA. El análisis inmunofenotípico es esencial porque las leucemias que no expresan mieloperoxidasa son las M0 LMA, M7 LMA y LLA.
El examen de los aspirados de médula ósea o de los especímenes de biopsia lo debe efectuar un oncólogo, un hematólogo, un hematopatólogo o patólogo general experto y capaz de interpretar especímenes convencionales y especialmente especímenes con tinciones.
Pronóstico y supervivenciaEntre los factores relacionados con los pronósticos en pacientes de LLA tenemos los siguientes:
- Edad: la edad, que es un factor significativo en la LLA infantil y en la LMA, también puede ser un factor pronóstico importante en la LLA de adultos. En un estudio, en general el pronóstico fue mejor en pacientes menores de 25 años; otro estudio encontró un mejor pronóstico para los pacientes menores de 35 años. Estos hallazgos pueden, en parte, estar relacionados con una mayor incidencia del Ph1 en pacientes de más edad que tienen LLA, un subgrupo relacionado con un pronóstico precario.[5,6]
- Compromiso del SNC: como en la LLA infantil, los pacientes adultos con LLA están en peligro de presentar complicaciones del sistema nervioso central (SNC) durante el curso de su enfermedad. Esto es especialmente cierto en aquellos pacientes con morfología L3 de (Burkitt).[7] Tanto el tratamiento como el pronóstico se ven afectados por esta complicación.
- Morfología celular: los pacientes con una morfología L3 muestran mejores resultados, según consta en el estudio Cancer and Leukemia Group B (CLB-9251[NCT00002494]), que ya se completó, cuando se les maneja de acuerdo a algoritmos de tratamiento específicos.[8,9] Este estudio descubrió que la leucemia L3 se pueden curar mediante el uso de regímenes quimioterapéuticos intensivos de ciclos rápidos tipo linfoma.[8,10,11]
- Anomalías cromosómicas: se han descrito anomalías cromosómicas como la aneuploidía y las traslocaciones que se pueden correlacionar con el pronóstico.[12] En particular, pacientes con leucemia linfoblástica aguda (LLA) positivos al cromosoma (Ph1) t(9;22) tienen un pronóstico precario y representan más del 30% de los casos adultos. Las leucemias Bcr-abl reordenadas que no muestran el clásico Ph1 presentan un pronóstico precario similar al de la LLA Ph1 positiva. Los pacientes con LLA Ph1 positiva son difíciles de curar con quimioterapia, aunque ahora se informa con cierta frecuencia de supervivencias a largo plazo cuando dichos pacientes se tratan con quimioterapia combinada e inhibidores de la tirosina cinasa Bcr-abl.
Otras dos anomalías cromosómicas con pronóstico precario son t(4;11), la cual se caracteriza por reordenamientos del gen MLL y que puede también ser reordenada a pesar de una citogenética normal y t(9;22). Además del t(9;22) y t(4;11), se ha informado que los pacientes con eliminación del cromosoma 7 o trisomía 8 tienen menos probabilidades de supervivencia a 5 años cuando se les compara con pacientes que presentan cariotipo normal.[13] En un análisis multivariado el cariotipo fue el factor pronóstico más importante de supervivencia sin enfermedad.[13][Grado de comprobación: 3iiDii]
Un seguimiento a largo plazo de 30 pacientes con LLA en remisión por al menos 10 años, ha mostrado 10 casos de cánceres secundarios. De 31 mujeres con supervivencia prolongada con LLA o leucemia mieloide aguda menores de 40 años de edad, 26 continuaron su período de menstruación normal después de terminar el tratamiento. Entre los 36 hijos de estas supervivientes que nacieron vivos, se presentaron dos problemas congénitos.[14]
Ensayos clínicos en cursoConsultar la lista del NCI de ensayos clínicos sobre el cáncer que se realizan en los Estados Unidos y que están aceptando pacientes. Para realizar la búsqueda, usar el término en inglés adult acute lymphoblastic leukemia. La lista de ensayos se puede reducir aun más por la ubicación donde se realizan, los medicamentos que se utilizan, el tipo de intervención y otros criterios. Nota: los resultados obtenidos solo estarán disponibles en inglés.
Asimismo, se dispone de información general sobre ensayos clínicos en el portal de Internet del NCI.
Bibliografía
- American Cancer Society.: Cancer Facts and Figures 2012. Atlanta, Ga: American Cancer Society, 2012. Available online. Last accessed July 31, 2012.
- Pui CH, Jeha S: New therapeutic strategies for the treatment of acute lymphoblastic leukaemia. Nat Rev Drug Discov 6 (2): 149-65, 2007. [PUBMED Abstract]
- Peterson LC, Bloomfield CD, Brunning RD: Blast crisis as an initial or terminal manifestation of chronic myeloid leukemia: a study of 28 patients. Am J Med 60(2): 209-220, 1976.
- Secker-Walker LM, Cooke HM, Browett PJ, et al.: Variable Philadelphia breakpoints and potential lineage restriction of bcr rearrangement in acute lymphoblastic leukemia. Blood 72 (2): 784-91, 1988. [PUBMED Abstract]
- Gaynor J, Chapman D, Little C, et al.: A cause-specific hazard rate analysis of prognostic factors among 199 adults with acute lymphoblastic leukemia: the Memorial Hospital experience since 1969. J Clin Oncol 6 (6): 1014-30, 1988. [PUBMED Abstract]
- Hoelzer D, Thiel E, Löffler H, et al.: Prognostic factors in a multicenter study for treatment of acute lymphoblastic leukemia in adults. Blood 71 (1): 123-31, 1988. [PUBMED Abstract]
- Kantarjian HM, Walters RS, Smith TL, et al.: Identification of risk groups for development of central nervous system leukemia in adults with acute lymphocytic leukemia. Blood 72 (5): 1784-9, 1988. [PUBMED Abstract]
- Lee EJ, Petroni GR, Schiffer CA, et al.: Brief-duration high-intensity chemotherapy for patients with small noncleaved-cell lymphoma or FAB L3 acute lymphocytic leukemia: results of cancer and leukemia group B study 9251. J Clin Oncol 19 (20): 4014-22, 2001. [PUBMED Abstract]
- Hoelzer D, Ludwig WD, Thiel E, et al.: Improved outcome in adult B-cell acute lymphoblastic leukemia. Blood 87 (2): 495-508, 1996. [PUBMED Abstract]
- Fenaux P, Lai JL, Miaux O, et al.: Burkitt cell acute leukaemia (L3 ALL) in adults: a report of 18 cases. Br J Haematol 71 (3): 371-6, 1989. [PUBMED Abstract]
- Reiter A, Schrappe M, Ludwig WD, et al.: Favorable outcome of B-cell acute lymphoblastic leukemia in childhood: a report of three consecutive studies of the BFM group. Blood 80 (10): 2471-8, 1992. [PUBMED Abstract]
- Chromosomal abnormalities and their clinical significance in acute lymphoblastic leukemia. Third International Workshop on Chromosomes in Leukemia. Cancer Res 43 (2): 868-73, 1983. [PUBMED Abstract]
- Wetzler M, Dodge RK, Mrózek K, et al.: Prospective karyotype analysis in adult acute lymphoblastic leukemia: the cancer and leukemia Group B experience. Blood 93 (11): 3983-93, 1999. [PUBMED Abstract]
- Micallef IN, Rohatiner AZ, Carter M, et al.: Long-term outcome of patients surviving for more than ten years following treatment for acute leukaemia. Br J Haematol 113 (2): 443-5, 2001. [PUBMED Abstract]
 Volver arriba
Volver arriba