Address correspondence to E.M. Faustman, University of Washington, Dept. of Environmental Health, 4225 Roosevelt Way NE, Suite 300, Seattle, WA 98105-6099. Telephone: (206) 685-2269. Fax: (206) 616-4875. E-mail: faustman@u.washington.edu
Although the research described in this article has been funded in part by the U.S. Environmental Protection Agency through R 826886-01-0 and the National Institute of Environmental Health Sciences through 1P01-ES09601-01 to E.M. Faustman, it has not been subjected to either agency's required peer and policy review and therefore does not necessarily reflect the views of either agency and no official endorsement should be inferred.
We thank T. Potter-Chiles for her outstanding administrative support and graphics expertise used in the preparation of this manuscript.
Received 3 May 1999; accepted 16 August 1999.
Identification, characterization, and control of environmental chemicals that adversely impact normal reproduction and development continue to be key public health goals. Approximately 250,000 U.S. children are born each year with birth defects diagnosed at or shortly after birth. Birth defects are the leading cause of infant mortality in the United States. Congenital anomalies, sudden infant death syndrome, and premature birth combined account for more than 50% of all infant mortality, yet the cause of most birth defects is unknown. Approximately 3-10% have been attributed to exogenous and environmental agents. Those environmental agents known to cause birth defects include lead; polychlorinated biphenyls; ethanol; organic mercury; and drugs such as thalidomide, diethylstilbestrol, valproic acid, and 13-
cis-retinoic acid. Most of these agents have been identified as developmental toxicants after tragic human exposures occurred (
1,2).
Up to 70% of all birth defects are of unknown etiology, and the role that environmental factors play in these occurrences is unknown. This lack of information impedes our ability to develop effective public health prevention strategies and has served as the foundation for increased public awareness and reinvigorated basic research on birth defects and children's health. When statistics about the number of years of potential lives lost are calculated for developmental effects and disabilities, these economic costs rival those for heart disease and cancer. Thus, identification of prevention pathways could alleviate not only social but also significant economic costs (3).
Purpose
The purpose of this article is to identify key factors that can help define the vulnerability of developing offspring, infants, and children to toxicant impacts and to give scientists clues for potential mechanisms that need to be evaluated to support the scientific basis for child-focused risk assessment. A focus of this article will be pesticides, given the heightened visibility of children and pesticide exposure following the 1993 National Academy of Sciences report titled "Pesticides in the Diets of Infants and Children" (4) and the specific focus of the recent Food Quality and Protection Act (5) . Environmental Health Perspectives recently published a special issue (6) on research newly funded under the Child Health grants program at the National Institute of Environmental Health Sciences and the U.S. Environmental Protection Agency (U.S. EPA) that provided an important opportunity to highlight examples of factors that need to be researched when assessing the potential susceptibility of children. This review uses the risk assessment paradigm for context, emphasizes examples of both in utero and postnatal developmental assessments, and introduces the related research aims of the University of Washington Center for Child Environmental Health Risks Research or Child Health Center.
Why Study Mechanisms of Susceptibility?
Risk is defined as the probability of adverse outcome and when applied to environmental health, risk assessment is usually defined as a function of both toxicity and exposure. Figure 1 shows a pictorial framework for child-specific risk characterization. To adequately evaluate the potential for enhanced health risks during development, scientists must consider child-specific factors affecting both toxicity and exposure. In the first section of this review, mechanisms of children's susceptibility relevant for toxicity assessment are identified and discussed. In the second section, examples of exposure factors that help to define children's susceptibility are presented. The final section discusses the implications of considering these factors for risk assessment
.

Figure 1. Framework for child-specific risk characterization.
Pesticides as Example Environmental Toxicants of Interest
Extensive use of pesticides to control insects and pests in order to protect human health and property and assist in food production has furthered public health worldwide. But many pesticides currently in use were registered by federal and state agencies before current toxicological testing protocols were developed or available. The effects of pesticides on children's learning and development have not received sufficient attention to date, and as a result there are few tests available to evaluate these behavioral end points. A National Academy of Sciences report (4) identified the lack of information as a public health issue and emphasized the importance of elucidating the health consequences of childhood exposures. The impacts of neurotoxicant exposure (e.g., pesticides) during development are of special interest because of the sensitivity of the developing nervous system to environmentally mediated toxicity; because normal nervous system functioning is essential to human activity, learning, and development; and because developmental effects will have consequences over an individual's entire life. On the U.S. EPA website (http://www.epa.gov/pesticides/), three key studies were discussed recently that re-emphasized the need to evaluate such chemicals for developmental neurotoxicity.
The complexity of normal development is reflected in the many integrated events that regulate cell growth, differentiation, and morphogenesis. Consequently, alterations at the molecular and cellular level that underlie subsequent developments may never be fully described. Of the myriad potential effects of environmental toxicants on these processes, scientists have focused their attention toward identification of critical or rate-limiting processes in which alterations would have significant manifestations in development, growth, and function. Wilson identified such critical processes as including altered mitosis, nucleic acid biosyntheses, membrane function/signal transduction, energy sources, inhibition of enzymes, and mutations (
7). Research has confirmed the ability of environmental chemicals to alter these processes/pathways and to produce significant adverse impacts on development. The significance of these alterations to human health must be put within a risk assessment framework in which factors such as dose and time of exposure are included in the overall characterization. This will allow public health decision makers to develop appropriate risk management strategies that minimize children's risk from the environment.
It is not the intent of this article to revisit all of the critical developmental pathways and the agents that disrupt them but to use example processes to illustrate child-specific factors that underlie susceptibility to environmental exposures. In this review, alterations in neuronal cell division, cell number, and differentiation are used as example cellular processes. Three factors are used to illustrate toxicity assessment considerations relevant for defining mechanisms of susceptibility--temporal, dose response, and genetic susceptibility considerations.
Illustration of Temporal Mechanisms of Susceptibility
Development of the human central nervous system (CNS) involves the production of 100 billion nerve cells and 1 trillion glial cells. Once produced, these neurons undergo migration, synaptogenesis, selective cell loss, and myelination. This development occurs unidirectionally, and inhibition at one developmental stage can cause alterations to subsequent processes (8,9). Figure 2 illustrates overall brain development by showing tissue hierarchy and neural origins for the ectodermally derived neural plate. Cell proliferation patterns are shown for specific example nuclei within the brain and demonstrate differences in the timing of peak periods of cell replication (Figure 2). Figure 3 outlines neurodevelopmental stages observed throughout the brain. These stages occur in temporally distinct time frames across different brain regions, making the brain heterogeneous in response to agents that interfere with these specific processes. For example, Figure 2 demonstrates the production of nerve cells in different regions of the CNS during gestation in the rat, and as shown, nuclei in the midbrain, cortex, and hippocampus undergo nerve cell production during distinct time frames. In addition to intraorgan differences in developmental processes, there may be substantial interspecies differences between rodents and humans in the development of the brain. Whereas in rodents the production of dentate gyrus granule cells of the hippocampus is largely conducted postnatally, the main production of these cells occurs prenatally in the human (10). Thus the behavioral and morphologic outcomes of gestational exposure to toxicants that cause cell death or that disrupt the cell cycle (e.g., cell-cycle inhibitors such as methyl mercury, taxol, vincristine, colchicine, 5-fluorouracil) would depend on dose and duration of exposure and the populations of potentially susceptible neuronal nuclei undergoing production at the time of exposure (11,12).

Figure 2: Illustration of overall rat brain development: tissue ontogeny and profiles of peak neuroepithelial cell proliferation within specific brain regions and nuclei throughout gestation. Figure adapted from Rodier et al. (11) with permission of John Wiley & Sons, Inc.

Figure 3. Illustration of overall brain development. This figure shows neurodevelopmental stages from the profile of neuroepithelial cell proliferation across brain regions (Figure 2) through processes of neuronal maturation (cell migration, differentiation, cell loss, and myelination) during normal brain development. Figure depicts pathway for neuronal maturation during normal brain development.
The historic use of organomercury-based fungicides and the present-day use of the benomyl and other benzimidazole analogues as anthelmintics are examples of cell-cycle inhibitors that have been investigated with regard to their influence on the developing CNS (13-17). Recent reports suggesting that chlorpyrifos (a pesticide used on many fruit crops such as apples, cherries, and pears) can cause selected brain cell loss also merit additional investigation (18,19).
Unlike other organ systems, the unidirectional nature of CNS development limits the capacity of the developing tissue to compensate for cell loss, and environmentally induced cell death can lead to a permanent reduction in cell number (20). Maintenance of this rigid temporal and spatial schedule allows the CNS to develop the morphologic characteristics associated with optimal function. It is this developmental complexity that appears to underlie the sensitivity of this organ to environmental influences and highlights the unique characteristics of development that make children at special risk from environmental exposures.
Identification of such critical processes is essential to our understanding of mechanisms of susceptibility. Thus, one of the laboratory-based research projects in the new Child Health Center at the University of Washington will focus on our understanding of the impacts of altering such critical neural processes and subsequent neurobehavioral function (21). Using three pesticides from different classes, these projects will use in vitro and in vivo rat systems to test the hypothesis that certain pesticides affect learning, growth, and development by altering the balance of cell proliferaion and cell death associated with development.
Dose-Response Considerations for Mechanisms of Susceptibility
The developing nervous system also provides an excellent illustration of the role of dose-response relationships in susceptibility. For example, classic studies with radiation exposures in the rat have shown steep dose-response relationships for brain malformations where a doubling of dose (50-100 rads) on day 9 of rodent gestation can cause a greater than 4-fold increase in rat brain malformations (9-41% incidence). At 200 rads, a 78% incidence of brain malformations was observed. On day 10, one day later in gestation, exposure to 50 rads does not produce brain malformations. Exposure to 100 rads produces only a 3% incidence, but exposure to 200 rads produces a 19% incidence. If exposure occurs earlier, on day 8, neither exposure to 50 nor 100 rads produces brain malformations (7). These observations convey the significance of evaluating both the dose and the timing of exposure to determine the stage and process of development that will be impacted. The relevancy of these observations is known for radiation exposure in humans, where irradiation of the human fetus at doses of 100 rads early in pregnancy can cause brain malformations such as microcephaly and mental retardation (22).
Cross-species comparison studies assessing methyl mercury exposure have been able to show consistent effects on neuronal cell number, differentiation, and morphologic organization at similar doses (22,23). In both examples, radiation and methyl mercury exposure altered cell number and cell division; these impacts have been postulated as modes of action for the observed adverse effects in neuronal development. The potential implications of such observations are evident when evaluated in context with research showing that altered cell proliferation and focal neuropathologic effects have been linked with specific neurobehavioral deficits (e.g., autism) (23-28).
A major emphasis of the molecular mechanisms project on our newly funded Child Health Center is on the evaluation of the potential impact of pesticides on neuronal cell replication and number, linking molecular events with in vivo neurobehavioral assessments. Frequently, the dose-response relationships established at a molecular/cellular level of assessment are inadequately linked with organ or whole-organism functional effects. Thus the significance of environmental contaminant-induced changes at the molecular level is inadequately utilized in risk assessment. It is a goal of our new Child Health Center to examine the links of all such changes with potential functional consequences.
Genetic Susceptibility
Genetic factors are also well recognized though poorly understood components contributing to individual variability in developmental responses to environmental exposures. Although associations linking specific genetic makeup (genotype) and enzyme function or protein activity (phenotype) to disease outcome have been explored extensively for cancer, research has been less focused on noncancer end points. However, where research has been conducted, it is clear that individual geno-phenotype can play a role in disease susceptibility (29,30).
Relevant for evaluating gene-environment relationships for developmental risk assessments are the known variations in drug metabolism enzymes associated with altered susceptibility to toxicant-induced birth defects. For example, extensive research by Nebert and colleagues (31) using early animal models has shown the importance of Ah receptor status and teratogenic response to benzo[a]pyrene, which requires metabolic activation by P450 enzymes. In human studies, the activity of the drug-metabolizing enzyme epoxide hydrolase has been linked with susceptibility of offspring to developmental toxicity following maternal exposure to diphenylhydantoin (32-34) and McCarver-May et al. (35) have shown that mothers who drink alcohol and have the alcohol dehydrogenase 2*3 allele are at lower risk of having a child with an adverse developmental outcome.
The examples described above demonstrate the importance of evaluating genetic variations in drug-metabolizing enzymes as genetic susceptibility factors important for developmental risk assessment. In addition to drug metabolism, however, genetic variability in growth factor regulators and homeobox genes may also underlie susceptibility. For example, an elevated risk of cleft palate has been reported for infants of mothers who smoke and carry an uncommon allele for transforming growth factor alpha (36-38). An increased risk of birth defects in smoking mothers has also been associated with a polymorphism in the homeobox genes (MSX) responsible for vertebrate limb development. Frequencies of rare alleles in the MSX1 locus were higher in infants with limb deficiencies when they were compared with infants with other types of birth defects. These risks were increased 4- to 5-fold when the infants who carry the rare alleles were from mothers who smoked during pregnancy (39).
Because individual susceptibility to environmental exposures likely involves specific genetic traits in combination with environmental exposures and other factors in order to produce disease, the nature of these multifactorial gene-environment interactions is likely to change throughout life. This is a particularly relevant question in regard to children's susceptibility to contaminants in their environment because of the relatively large changes in gene expression that occur during development as well as the dynamic nature of their changing lifestyle exposures.
A good example of this temporal sensitivity in gene-environment interaction is seen with methemoglobinemia, where both a temporal and genetic susceptibility component must be evaluated. Ingested nitrates can be converted to nitrites by intestinal bacteria, and nitrite ions can bind with hemoglobin, forming methemoglobin, a form of hemoglobin with reduced oxygen-carrying capacity. This reduction in oxygen capacity can result in anemia or "blue baby syndrome" in infants who have fetal forms of hemoglobin and less functional methemoglobin reductase (MR). MR is an enzyme that normally reduces methemoglobin back to its oxygen-carrying state in adults and older children. The enhanced susceptibility of very young infants lacking or having minimally functional MR to methemoglobemia has been of concern for infants fed formula prepared with water contaminated with high agricultural runoff from nitrate fertilizers, as it has been estimated that 40% of applied nitrates can enter water sources as field runoff/leachate (40). In addition to environmental sources, chronic congenital methemoglobinemias may also result from an inherited deficiency of MR or abnormal amino acid substitution in globin M and H chains (41).
An example of a potentially important genetic susceptibility marker in pesticide neurotoxicology is the polymorphism in the paraoxonase (Pon 1) gene. This enzyme metabolizes and inactivates a wide variety of organophosphate (OP) oxons, the active forms of OP pesticides, including methyl parathion, which was recently severely restricted by the U.S. EPA. By design, OP pesticides inactivate acetylcholinesterase (AchE)-mediated metabolism of acetylcholine (ACh) by impairing AchE central and peripheral nervous system activity.
The human Pon 1 gene has been cloned and shown to have a polymorphism at position 192. When arginine (Arg192) is present at this position, Pon 1 hydrolyzes the OP paraoxon rapidly; however, when this site is replaced with glutamine (Gln192), paraoxon is hydrolyzed more slowly (42,43).
The available research in this area demonstrates a strong need to ascertain individual geno-phenotype in which both genetic polymorphism status is determined and the enzyme activity is measured against varied OP substrates. If an individual is homozygous for the low-activity allele (Gln192) and has low Pon1 expression levels, then the individual would hydrolyze both paraoxon and the oxon of chlorpyrifos very slowly. Such an individual would be predicted to be more susceptible to the effects of both these OP pesticides than a comparable individual with high Pon1 expression (44). Whereas the activity of the enzyme is high toward paraoxon when arginine is at position 192 and low when glutamine is at this position, the situation is reversed when the substrate is diazoxon, soman, or sarin, demonstrating the need for genotyping information for predictive risk assessment (44).
Young animals are more sensitive to the acute toxic effects of OPs than adult animals (45-49). Brain ACh levels fail to account for these differences, but metabolic studies have suggested the differences may be due to an age-related decrease in paraoxonase activity in human infants and children. Levels of Pon 1 activity in rodents do not reach adult levels until 4 weeks of postnatal development (50). L. Costa and C. Furlong will direct a project in the Child Health Center using Pon 1 knockout mice to ascertain both the potential age and substrate susceptibility of OP-induced developmental neurotoxicity and will link these biochemical effects with functional neurobehavioral impacts. The significance for endogenous ACh metabolism and pesticide toxicity is unknown. Gaining knowledge about the age and genetic differences in this enzyme is especially significant given the role that the cholinergic system plays in learning and memory (51) and the new findings that suggest a role for ACh in various aspects of normal brain development (52-54). The Child Health Center's paraoxonase polymorphism study will address this research need.
Three examples of the types of exposure considerations needed for evaluating children's risks include
a) differences in types of exposures,
b) unique pathways of exposure, and
c) child-specific toxicokinetics.
Types and Frequency of Exposures
Numerous reviews and discussions of differences in the types of exposures that children receive have been published (4,55,56). In this section, several examples will be used to illustrate two key points: the unique differences in types of exposures that children may experience during each developmental phase, and the dynamic nature of these exposure patterns.
Figures 4 and 5 show examples of the types and quantity of dietary exposure from specific food sources. This information was prepared from a survey conducted in the United Kingdom by Mills and Tyler in 1992 (56). This survey found that solid foods consisting of cereals (baby rice or rusks) were introduced to infants at approximately 13 weeks of age. Up to 20% of the infants in the study were fed pureed fruits or vegetables, and ingestion of fruit juices was common. By 6 months of age, approximately 60% of the infants were reported to be ingesting at least some of the same basic foods as the family. For risk assessment purposes, such differences over time in exposure profiles suggest distinct exposure subgroups even within early child development. These findings also suggest that if adverse health impacts of an environmental contaminant are suspected, the identification of such impacts would be very difficult without detailed knowledge of the exposure patterns within these subgroups.
 |
Figure 4. Age-related consumption of foods and beverages. Data from Lawrie (55) with permission of Taylor & Francis.
|
 |
Figure 5. Age-related consumption of foods and beverages as a ratio of intake to body weight. Data from Lawrie (55) with permission of Taylor & Francis.
|
A unique exposure pathway of concern for exposure of infants to environmental toxicants during their first year and a half of life is the consumption of breast milk. Figures 4 and 5 show the relationship of breast milk in comparison to other food sources. Breast milk can be a source of exposure for infants that not only reflects a subset of maternal food consumption exposures (primarily lipid extractable constituents) but also maternal exposures to an array of exogenous chemical compounds including drugs, occupational and environmental metals, solvents, and pesticides (57-62). Analyses of breast milk residue that have focused on fat-soluble residues have demonstrated the presence of polyhalogenated biphenyls, dibenzodioxins, dibenzofurans, and the organochlorine pesticides (e.g., DDT), lindane, hexachlorocyclohexane, dieldrin) in milk from mothers worldwide, reflecting global transport of these compounds (57,63-69). Recognition that many of these compounds are environmentally persistent has resulted in regulatory controls and in generally declining residue levels in humans for polychlorinated biphenyls and DDT.
The partitioning of environmental compounds to breast milk is strongly associated with the affinity of these chemicals for milk lipids. In some cases, such as for the organochlorine compounds, the half-life of these compounds in breast milk can be extremely long (70). Breast-feeding is a principal means of reducing maternal burden but at the expense of exposure to the infant (70-72). Because of the extremely long half-life of these compounds in the mother, fetal exposure can derive from a maternal body burden accumulated over a period of many years. Thus exposure estimates based on default exposure assumptions can be problematic. Direct biomonitoring of breast milk is the most useful metric for risk estimation. However, because of the complexities of the kinetics of accumulation, this biomonitoring provides limited clues for ascertaining specific maternal exposure patterns, as it presents an average estimate of accumulated maternal exposures.
Despite the potential risks associated with developmental exposure to environmental contaminants in breast milk, breast milk also provides a host of nutritional factors associated with immunologic and psychologic development and general health. Limited formal quantitative analyses have been performed to evaluate the net health effects of breast-feeding. However, the health benefits associated with breast feeding have generally been evaluated as outweighing the health risks associated with most environmental contaminants, with exceptions for some therapeutics and illicit drugs (4,59,73-76).
Exposure Pathways--Age-Specific Behaviors
Children face potentially elevated risk of pesticide-induced toxicity due to age-specific behaviors that can increase exposure. Younger children have distinct dietary patterns, both in terms of food selection and amounts consumed (4). Younger children also routinely explore their environment, putting fingers, toys, and other objects into their mouths. Hand contact with floors, carpet, lawns, and other surfaces during crawling may lead to enhanced exposure via hand-to-mouth and object-to-mouth transfer. A subset of children exhibit pica (deliberate ingestion of nonfood items). In particular, soil ingestion rates of geophagic (dirt-specific forms of pica) children may be dramatically elevated, although they are currently poorly defined (77). Children's skin contact with surfaces via crawling or play may also contribute to exposure to environmental compounds even in the absence of ingestion. The dermal availability of many pesticides and the importance of the dermal pathway with respect to occupational exposures in agriculture are well established (78). Because children have higher ratios of skin surface area to body weight (roughly double in infancy) than adults and probably experience more intensive contact with their surroundings than adults in nonoccupational settings, increased susceptibility to dermal absorption of contaminants is plausible. Limited data from an investigation of inappropriate residential use of methyl parathion suggest an association between surface contamination and elevated levels of pesticide metabolite in the urine of children under 3 years of age (79). However, the relative contributions of dermal absorption and ingestion cannot be ascertained from the information available.
In agricultural communities where pesticides are used in crop production, children have the potential for exposure through additional pathways. If children live in close proximity to pesticide-treated farmland, for example, they may contact pesticides through normal play in and around the home. Children in agricultural communities can also be exposed to workplace pesticides if parents bring in these chemicals on clothing or skin. This pathway is sometimes referred to as take-home exposure (80).
Since 1992 several studies at the University of Washington Pacific Northwest Agricultural Safety and Health (PNASH) Center (Seattle, WA) have focused their research efforts on children's exposure to OP pesticides in the agricultural regions of Washington State (81,82). The first of these studies examined pesticides in soil and house dust. Concentrations of four OP pesticides were substantially higher in the soil and house dust at residences of so-called agricultural families (families with a parent working with pesticides in agriculture) than at the residences of reference families (home at least one-quarter of a mile from treated farmland and no family member working in agriculture) (81). A subsequent study focused on biological monitoring (urine sampling) of children in the same community and found that children of pesticide applicators had median OP pesticide metabolite concentrations that were 4 times higher than those of reference children (82). Thus, both proximity to agricultural pesticide use and parental take-home of pesticides appear to contribute to elevated body burdens in young children in this community. When urinary metabolites were converted to pesticide dose estimates for the OP pesticide azinphos-methyl, 53% of these doses exceeded the U.S. EPA chronic reference dose of 0.015 µg/kg (83). Recently, the U.S. EPA has taken action to restrict azinphos-methyl use as a step to protect children's health by reducing exposure. These results are consistent with results of studies looking at other occupational take-home exposure pathways. Several studies have shown that at-home exposures of children or family members result from a parent worker being exposed to lead or asbestos (84-87). Recently, homes and vehicles of lead-exposed workers were found to have higher levels of lead contamination than similar homes and vehicles of families with no take-home pathway (88,89). This evidence supports the idea that contamination of a worker's skin or clothing can lead to elevated doses in children with whom they live.
As part of the Child Health Center, a community intervention study to reduce take-home pesticide exposure among agricultural field workers is planned as a collaboration between the Fred Hutchinson Cancer Research Center (Seattle, WA) and the PNASH Center. The study will take place in the lower Yakima Valley, one of the most productive agricultural regions in Washington State. Interviews, environmental samples, and biological samples will be collected in the first and fourth years of the study. In the second and third year, community-based intervention strategies designed to break the take-home exposure pathway will be implemented in 14 communities. An additional 14 communities will serve as controls.
The role of pesticide drift as a contributor to children's exposure will also be the subject of investigation. With support from the Child Health Center and the PNASH Center, researchers will utilize two novel techniques for characterizing exposure associated with pesticide drift. Global positioning system technology will be used to create detailed spatial and temporal maps of children's activities. A portable light detection and ranging (LIDAR) system will be used to measure particle concentration in spray plumes generated by airblast orchard applications, and air sampling on the ground will provide pesticide concentration data needed to calibrate the LIDAR system. The combination of these data will allow analysis of drift exposure for children with a high degree of temporal and spatial resolution. Simultaneous biological monitoring will allow testing of exposure and dose models based on environmental modeling.
Toxicokinetic Considerations
Important factors that define children's susceptibility to contaminants are the age-related toxicokinetic changes in development. Toxicokinetics include evaluation of the four processes of absorption, distribution, biotransformation, and excretion of toxicants. In particular, it describes the rate of action of these processes on contaminants and allows the risk assessor to link exposure with the amount, duration, and form of the toxicant interactions with the target organs. For purposes of this review, this section of the paper focuses on several example changes occurring during pregnancy and early childhood that can affect the doses that children and their tissues receive.
During pregnancy, many physiologic changes occur, and these changes can impact the kinetic aspects of chemicals (90). For example, up to an 85% change in plasma flow to the kidney has been observed during pregnancy. This dramatic increase can facilitate elimination of many compounds normally removed via renal excretion and minimize the amount of such compounds that could reach the developing conceptus. Blood albumin concentrations can decrease during pregnancy and have been reported to be lowered by up to 30%. This decrease in plasma proteins could result in a change of the ratio of chemical bound to plasma protein versus free compound, thus altering the amount of free compound available for transport across membranes into specific organs. In all cases, time-specific information is needed to estimate the significance of these changes for conceptal exposures, as these physiologic changes occur with different rates throughout the course of pregnancy.
Of particular interest in discussion of mechanisms of susceptibility relevant for toxicant exposure are kinetic conditions under which the embryo/fetus would experience greater concentrations of a contaminant than the mother. Defining "protective" levels of exposure in the mother would not necessarily translate to protective levels for the conceptus. This can occur when the in utero compartment acts as a deep compartment. Compounds that accumulate in fetal organs include heavy metals, DDT, polyhalogenated biphenyls, and tetracycline (90).
Although both fetal and maternal biotransformation capabilities are important, the ability of the conceptus and neonate to biotransform chemicals to reactive metabolites is especially relevant for our considerations for two reasons. First, if the fetus metabolizes a compound to a more charged, less polar metabolite, this metabolite could be kinetically hindered from crossing back across membranes to be eliminated by the maternal elimination pathways. Thus, metabolite concentrations could build up within the fetal compartment. The other significant consideration with embryonic/fetal metabolism is the potential to generate reactive metabolites within conceptal tissues with less well-developed protective/conjugative pathways.
Figure 6 taken from Cresteil (91) shows elegant work to evaluate the evolution of various isoforms of human liver cytochrome P450s in utero and throughout childhood. As is evident from this figure, each isoform has its own developmental profile. Given that each isoform has its own organ and substrate preference profiles, the complexity of predicting metabolite profiles for a given compound or mixture is extremely challenging. Even more challenging is using this information for predicting human conceptal and newborn risks for single agents or complex mixtures. Evidence on the possible role of genetic polymorphisms of drug-metabolizing enzymes for human risk assessment was discussed in an earlier section of this paper on genetic susceptibility.

Figure 6. Evolution of cytochrome P450 isoforms in the human liver during growth in humans. Data from Cresteil (91) with permission of Taylor & Francis.
The adage that "children are not just small adults" has implications for establishing safe levels of exposure for pesticides. This means that exposure criteria should be based on information relevant to predicting risks to children and should account for such toxicokinetic differences occurring with development. These issues are partially recognized in pharmaceutic dosing recommendations through the use of allometric scaling to account for age-related changes in body composition and activity. However, reliance on such practices must be performed along with consideration of chemical-specific characteristics that influence both target organ exposure and potency. Presented in Figures 7 and 8 are examples of changes in body composition (i.e., weight and surface area) during the first 17 years of life, both of which are commonly used as scaling factors in pharmacologic dosing determinations.
 |
Figure 7. Age-dependent changes in surface area. Data adapted from Renwick (92).
|
 |
Figure 8. Age-dependent changes in body weight (kg) or weight/surface area (kg/m2). These (and other) body composition metrics are commonly used to scale exposures across age groups for pharmaceuticals. Data adapted from Renwick (91).
|
Lack of consideration of toxicokinetic and toxicodynamic factors in establishing exposure criteria for pesticide can lead to errors in risk models. For example, if pesticide exposure tolerance values are based on adults and if pharmacokinetic parameters are linearly scaled by weight, then a factor of 17.6 should be used, whereas a factor of 8.2 should be used if exposure should be linearly scaled by surface area. Other allometric scaling factors might be better described using total body water, percentage intracellular water, total body protein, total bone mineral, or other factors. Clearly, the most fully informed decision will have detailed information regarding the driving factors influencing absorption, distribution, metabolism and elimination and how they change with age (92).
Implications for Risk Assessment
The idea that "children are not just small adults" has been reiterated in many recent documents following the National Research Council's 1993 report on "Pesticides in the Diets of Infants and Children" (
4), yet we have continued to conduct risk assessments for environmental agents in isolation of this fact. This is due to several factors. First, the scientific basis on how, when, and by how much children differ from adults in their susceptibility to environmental insults is inadequate. This lack of scientific information was highlighted in the 1997 National Research Conference on Children's Environmental Health: Research, Practice, Prevention, Policy report (
93) that made important recommendations for research to address these deficiencies. In addition, risk assessment methods have not yet been designed to utilize child-specific susceptibility information to improve public health evaluations even when such information is available. Hence, we rely on 10-fold safety factors applied to the reference dose for pesticides with suspected effects on children when the true differences between children and adult susceptibility may range over multiple orders of magnitude.
In this review we have illustrated examples of mechanisms that underlie the susceptibility of children to environmental exposures that are relevant for both toxicity and exposure assessment. Such mechanisms must be put into the context of the risk assessment framework to be tied to effective risk management. Failure to do this will result in continued discussion of the relevancy of factors for children's risk with little progress made toward improving children's health.
In this paper we discussed mechanisms of susceptibility for toxicity assessment and highlighted the need to identify critical pathways of normal development that might be at heightened risk for susceptibility to toxicant impacts. An example discussed was neurons in specific brain regions undergoing select patterns of proliferation and differentiation. On a molecular and cellular level we can, and will, measure these effects as part of research in our Child Health Center. However, for risk assessment these observations need to be drawn into a risk assessment context. What health impacts result when 10, 15, or 20% of neurons are lost on day 13 in rodent midbrains following exposure to a toxicant? To answer this question, the functional consequences of such loss must be evaluated and is the underlying rationale for our planned studies to link molecular and cellular changes with altered neurobehavioral assessment. Once this functional link is established, exposure assessments must allow us to link these functional effects with environmental exposures. As discussed in this review, the complexities of environmental exposure are great, challenging the risk assessor both to understand the biological significance of specific exposures at selected times throughout gestation and to evaluate chronic low-dose exposure over extended developmental processes. Linkage of each of these types of exposures with functional impacts is needed. To accomplish this we have developed linked models of assessment where exposure, toxicokinetics, and toxicodynamics can be considered in the same risk models. Figure 9 illustrates such a biologically based dose-response assessment approach. In this model, developed for methyl mercury and being expanded for other toxicants/pesticides, we have unified our risk assessment process (94,95). The challenge to our center investigators is to develop similar unifying approaches for our molecular/cellular research that are linked with functional assessments and with relevant exposure assessment.

Figure 9. Examples of a limited TK and TD model for developmental risk assessment. Toxicodynamic, TD; toxicokinetic, TK. In this example a hypothesized TD model is shown to describe molecular and cellular events within the conceptal brain. This TD model is linked with a hypothetical TK model describing toxicant delivery to the brain during development. It serves as an example of the need to link both exposure and molecular effects to predict health risks [For more details and explanation, please refer to Faustman et al. (94)].
Finally, by framing the available information in a risk assessment context, both scientists and regulators will continue the process of putting relevant science into risk assessment. As indicated in the National Academy of Sciences framework, only through the iterative loop between basic research and risk assessment can the informational needs of public health decision makers be met. Moreover, by using the biologically based risk assessment models to generate hypotheses regarding factors that may strongly influence child health risks, this information can be used to prioritize research. An example of the need for improved child-specific risk assessment research is the recent U.S. EPA-announced restriction on the use of two organopesticides that are used commonly on produce regularly eaten by children, methyl parathion and azinphos-methyl (Guthion). As we have demonstrated in this review, there are many reasons to believe that children may have unique sensitivities to their environment. The health implications of these sensitivities can only be understood through continued research.
REFERENCES AND NOTES
1. Shepard T. Catalog of Teratogenic Agents. Baltimore, MD: Johns Hopkins University Press, 1989.
2. Schardein J. Chemically Induced Birth Defects. New York:Marcel Dekker, 1985.
3. Morbidity and Mortality Weekly Report. Economic Costs of Birth Defects and Cerebral Palsy-United States, 1992. Centers for Disease Control 44:694-699 (1995).
4. National Research Council. Pesticides in the Diets of Infants and Children. Washington, DC:National Academy Press, 1993.
5. Food Quality Protection Act of 1996. Public Law 104-170. 104th Congress, 1996.
6. Dearry A, Collman G, eds. Children's Environmental Health and Disease Prevention Research. Environ Health Perspect 107(suppl 3):389-460 (1999).
7. Wilson J. Current status of teratology: general principles and mechanisms derived from animal studies. In: Handbook of Teratology: General Principles and Etiology (Wilson J, Fraser F, eds). New York:Plenum Press, 1977;47.
8. Herschkowitz N. Brain development in the fetus, neonate and infant. Biol Neonate 54:1-19 (1988).
9. Langman J, Webster W, Rodier P. Morphological and behavioural abnormalities caused by insults to the CNS in the perinatal period. In: Teratology: Trends and Applications (Berry CL, Poswillo DE, eds). New York:Springer-Verlag, 1975;182-200.
10. Bayer SA, Altman J, Russo RJ, Zhang X. Timetables of neurogenesis in the human brain based on experimentally determined patterns in the rat. Neurotoxicology 14:82-144 (1993).
11. Rodier P. Correlations between prenatally-induced alterations in CNS populations and postnatal function. Teratology 16:235-246 (1977).
12. Rodier P. Chronology of neuron development: animal studies and their clinical implications. Dev Med Child Neurol 22:525-545 (1980).
13. Ponce R, Kavanagh T, Mottet N, Whittaker S, Faustman E. Effects of methyl mercury on the cell cycle of primary rat CNS cells in vitro. Toxicol Appl Pharmacol 127:83-90 (1994).
14. Sager P, Aschner M, Rodier P. Persistent, differential alterations in developing cerebellar cortex of male and female mice after methylmercury exposure. Brain Res 314:1-11 (1984).
15. Clarkson T, Amin-Zaki L, Al-Tikriti S. An outbreak of methylmercury poisoning due to consumption of contaminated grain. Fed Proc 35:2395-2399 (1976).
16. Hoogenboom ER, Ransdell J, Ellis W, Kavlock R, Zeman F. Effects on the fetal rat eye of maternal benomyl exposure and protein malnutrition. Curr Eye Res 10:601-612 (1991).
17. Kavlock R, Chernoff N, Gray L Jr, Gray J, Whitehouse D. Teratogenic effects of benomyl in the Wistar rat and CD-1 mouse, with emphasis on the route of administration. Toxicol Appl Pharmacol 62:22-54 (1982).
18. Campbell C, Seidler F, Slotkin T. Chlorpyrifos interferes with cell development in rat brain regions. Brain Res Bull 43:179-189 (1977).
19. Cosenza M, Bidanset J. Effects of chlorpyrifos on neuronal development in rat embryo midbrain micromass cultures. Vet Hum Toxicol 37:118-121 (1995).
20. Bayer S. Cellular aspects of brain development. Neurotoxicology 10:307-320 (1989).
21. The University of Washington Center for Child Environmental Health Risks Research's Molecular Mechanisms Study and Neurobehavioral Assessment core are directed by P. Mirkes and T. Burbacher, respectively.
22. Mettler F, Upton A. Medical Effects of Ionizing Radiation. Philadelphia:W.B. Saunders, 1995.
23. Burbacher T, Rodier P, Weiss B. Methylmercury developmental neurotoxicity: a comparison of effects in humans and animals. Neurotoxicol Teratol 12:191-202 (1990).
24. Rodier P. Differential structural effects of three behavioral teratogens. Dev Toxicol Environ Sci 11:53-60 (1983).
25. Rodier P, Aschner M, Sager P. Mitotic arrest in the developing CNS after prenatal exposure to methylmercury. Neurobehav Toxicol Teratol 6:379-385 (1984).
26. Schull W, Norton S, Jensh R. Ionizing radiation and the developing brain. Neurotoxicol Teratol 12:249-260 (1990).
27. Wallace R, Altman J. Behavioral effects of neonatal irradiation of the cerebellum. II: Qualitative studies in young-adult and adult rats.op Psychobiol 2:266-272 (1970).
28. Wallace R, Altman J. Behavioral effects of neonatal irradiation of the cerebellum. I: Qualitative observation before and after weaning. Develop Psychobiol 2:257-265 (1970).
29. Costa L, Omiecinski C, Faustman E, Omenn G. Ecogentics: determining susceptibility to chemical-induced diseases. Washington Public Health 11:8-11 (1993).
30. Omenn G, Omiecinski C, Eaton D. Eco-genetics of chemical carcinogens. In: Biotechnology and Human Genetic Predisposition to Disease (Cantor C, Caskey C, Hood L, eds). New York:Wiley-Liss, 1990;81-93.
31. Shum S, Jensen NM, Nebert DW. The murine Ah locus: in utero toxicity and teratogenesis associated with genetic differences in benzo[a]pyrenemetabolism. Teratology 20:365-376 (1979).
32. Strickler SM, Dansky LV, Miller MA, Seni MH, Andermann E, Spielberg SP. Genetic predisposition to phenytoin-induced birth defects. Lancet 2:746-749 (1985).
33. Buehler BA, Delimont D, van Waes M, Finnell RH. Prenatal prediction of risk of the fetal hydantoin syndrome [See Comments]. N Engl J Med 322:1567-1572 (1990).
34. Spielberg SP. Pharmacogenetics and the fetus [Editorial]. N Engl J Med 307:115-116 (1982).
35. McCarver-May DG, Thomasson H, Martier SS, Sokol R, Li T-K. Presence of the ADH2*3 gene protects against neurobehavioral deficits in offspring exposed to intrauterine ethanol. J Pharmacol Exp Ther 283:1095-1101 (1997).
36. Shaw GM, Lammer EJ. Incorporating molecular genetic variation and environmental exposures into epidemiologic studies of congenital anomalies. Reprod Toxicol 11:275-280 (1997).
37. Shaw GM, Wasserman CR, Lammer EJ, O'Malley CD, Murray JC, Basart AM, Tolarova MM. Orofacial clefts, parental cigarette smoking, and transforming growth factor-alpha gene variants. Am J Hum Genet 58:551-561 (1996).
38. Hwang SJ, Beaty TH, Panny SR, Street NA, Joseph JM, Gordon S, McIntosh I, Francomano CA. Association study of transforming growth factor alpha (TGF 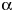 ) TaqI polymorphism and oral clefts: indication of gene-environment interaction in a population-based sample of infants with birth defects. Am J Epidemiol 141:629-636 (1995).
) TaqI polymorphism and oral clefts: indication of gene-environment interaction in a population-based sample of infants with birth defects. Am J Epidemiol 141:629-636 (1995).
39. Hwang SJ, Beaty TH, McIntosh I, Hefferon T, Panny SR. Association between homeobox-containing gene MSX1 and the occurrence of limb deficiency. Am J Med Genet 75:419-423 (1998).
40. Levi P. Toxicity of chemicals. In: A Textbook of Modern Toxicology (Hodgson E, Levi P, eds). New York:Elsevier, 1987;185-232.
41. Smith R. Toxic responses of the blood. In: Casarett and Doull's Toxicology: The Basic Sciences of Poisons (Klaassen D, Amdur M, Doull J, eds). New York:McGraw-Hill, 1996;335-354.
42. Humbert R, Adler DA, Disteche CM, Hassett C, Omiecinski CJ, Furlong CE. The molecular basis of the human serum paraoxonase activity polymorphism. Nat Genet 3:73-76 (1993).
43. Hassett C, Richter R, Humbert R, Chapline C, Crabb J, Omiecinski C, Furlong C. Characterization of cDNA clones encoding rabbit and human serum paraoxonase: the mature protein retains its signal sequence. Biochemistry 30:10141-149 (1991).
44. Davies HG, Richter RJ, Keifer M, Broomfield CA, Sowalla J, Furlong CE. The effect of the human serum paraoxonase polymorphism is reversed with diazoxon, soman and sarin. Nat Genet 14:334-336 (1996).
45. Brodeur J, DeBois K. Comparison of acute toxicity of anticholinesterase insecticides to weanling and adult male rats. Proc Soc Exp Biol Med 114:509-511 (1963).
46. Lu F, Jessup D, Lavallee A. Toxicity of pesticides in young versus adult rats. Food Cosmet Toxicol 3:591-596 (1965).
47. Benke G, Murphy S. The influence of age on the toxicity and metabolism of methyl parathion and parathion in male and female rats. Toxicol Appl Pharmacol 31:254-269 (1975).
48. Harbison R. Comparative toxicity of some selected pesticides in neonatal and adult rats. Toxicol Appl Pharmacol 32:443-446 (1975).
49. Pope C, Chakraborti T, Chapman M, Farrar J, Arthun D. Comparison of in vivo cholinesterase inhibition in neonatal and adult rats by three organophosphorothioate insecticides. Toxicology 68:51-61 (1991).
50. Li W, Matthews C, Disteche C, Costa L, Furlong C. Paraoxonase (PON1) gene in mice: sequencing, chromosomal localization and developmental expression. Pharmacogenetics 7:137-144 (1997).
51. Everitt B, Robbins T. Central cholinergic systems and cognition. Annu Rev Psychol 48:649-684 (1997).
52. Costa L. Muscarinic receptors in the developing nervous system. In: Receptors in the Developing Nervous System (Zagon I, McLoughlin P, eds). London:Chapman and Hall, 1993;21-42.
53. Guizzetti M, Costa P, Peters J, Costa L. Acetylcholine as a mitogen: muscarinic receptor-mediated proliferation of rat astrocytes and human astrocytoma cells. Eur J Pharmacol 297:265-273 (1996).
54. Yan G, Lin S, Irwin R, Paul S. Activation of muscarinic cholinergic receptors blocks apoptosis of cultured cerebellar granule neurons. Mol Pharmacol 47:248-257 (1995).
55. Lawrie C. Different dietary patterns in relation to age and the consequences for intake of food chemicals. Food Addit Contam 15:75-81 (1998).
56. Mills A, Tyler H. Food and nutrient intakes of British infants aged 6-12 months. London:Her Majesty's Stationery Office, 1992.
57. Quinsey P, Donohue D, Ahokas J. Persistence of organochlorines in breast milk of women in Victoria, Australia. Food Chem Toxicol 33:49-56 (1995).
58. Stabin MG. Health concerns related to radiation exposure of the female nuclear medicine patient. Environ Health Perspect 105(suppl 6):1403-1409(1997).
59. Rogan W. Pollutants in breast milk . Arch Pediatr Adolesc Med 150:981-990 (1996). [Published erratum appears in Arch Pediatr Adolesc Med 150(12):1282 (1996).
60. Howard C, Lawrence R. Breast-feeding and drug exposure. Obstet Gynecol Clin North Am 25:195-217 (1998).
61. Abadin H, Hibbs B, Pohl H. Breast-feeding exposure of infants to cadmium, lead, and mercury: a public health viewpoint. Toxicol Ind Health 13:495-517 (1997).
62. Wolff MS. Occupationally derived chemicals in breast milk. Am J Ind Med 4:259-281 (1983).
63. Lutter C, Iyengar V, Barnes R, Chuvakova T, Kazbekova G, Sharmanov T. Breast milk contamination in Kazakhstan: implications for infant feeding. Chemosphere 37:1761-1772 (1998).
64. Beck H, Dross A, Mathar W. PCDD and PCDF exposure and levels in humans in Germany. Environ Health Perspect 102(suppl 1):173-185 (1994).
65. Duarte-Davidson R, Jones K. Polychlorinated biphenyls (PCBs) in the UK population: estimated intake, exposure and body burden. Sci Total Environ 151:131-152 (1994).
66. Feeley M. Biomarkers for Great Lakes priority contaminants: halogenated aromatic hydrocarbons. Environ Health Perspect 103(suppl 9):7-16 (1995).
67. Siddiqui M, Saxena M, Bhargava A, Seth T, Murti C, Kutty D. Agrochemicals in the maternal blood, milk, and cord blood: a source of toxicants for prenates and neonates. Environ Res 24:24-32 (1981).
68. Dewailly E, Ayotte P, Laliberte C, Weber J, Gingras S, Nantel A. Polychlorinated biphenyl (PCB) and dichlorodiphenyl dichloroethylene (DDE) concentrations in the breast milk of women in Quebec [See Comments]. Am J Public Health 86:1241-1246 (1996).
69. Dewailly E, Nantel A, Weber J, Meyer F. High levels of PCBs in breast milk of Inuit women from arctic Quebec. Bull Environ Contam Toxicol 43:641-646 (1989).
70. Kreuzer P, Csanady G, Baur C, Kessler W, Papke O, Greim H, Filser J. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) and congeners in infants. A toxicokinetic model of human lifetime body burden by TCDD with special emphasis on its uptake by nutrition. Arch Toxicol 71:383-400 (1997).
71. Abraham K, Papke O, Gross A, Kordonouri O, Wiegand S, Wahn U, Helge H. Time course of PCDD/PCDF/PCB concentrations in breast-feeding mothers and their infants. Chemosphere 37:1731-1741 (1998).
72. Schecter A, Kassis I, Papke O. Partitioning of dioxins, dibenzofurans, and coplanar PCBS in blood, milk, adipose tissue, placenta and cord blood from five American women. Chemosphere 37:1817-1823 (1998).
73. Scialli A. A Clinical Guide to Reproductive and Developmental Toxicology. Boca Raton, FL:CRC Press, 1992.
74. Kacew S. Current issues in lactation: advantages, environment, silicone. Biomed Environ Sci 7:307-19(1994).
75. Wormworth J. Toxins and tradition: the impact of food-chain contamination on the Inuit of northern Quebec. Cmaj 152:1237-40(1995).
76. Korrick S, Altshul L. High breast milk levels of polychlorinated biphenyls (PCBs) among four women living adjacent to a PCB-contaminated waste site. Environ Health Perspect 106:513-518 (1998).
77. U.S. EPA. Aggregate Exposure. Review Document for the Scientific Advisory Panel SAP Public Docket. Washington, DC: U.S. Environmental Protection Agency, 1997
78. Fenske R. Pesticide exposure assessment of workers and their families. Occup Med 12:221-237 (1997).
79. Esteban E, Rubin C, Hill R, Olson D, Pearce K. Association between indoor residential contamination with methyl parathion and urinary para-nitrophenol. J Expo Anal Environ Epidemiol 6:375-387 (1996).
80. NIOSH. Report to Congress on Workers' Home Contamination Study Conducted Under the Workers Family Protection Act (29 U.S.C. 671a). Cincinnati, OH:National Institute for Occupational and Safety and Health, 1995.
81. Simcox N, Fenske R, Wolz S, Lee I, Kalman D. Pesticides in household dust and soil: exposure pathways for children of agricultural families. Environ Health Perspect 103:1126-1134 (1995).
82. Loewenherz C, Fenske R, Simcox N, Bellamy G, Kalman D. Biological monitoring of organophosphorus pesticide exposure among children of agricultural workers in central Washington State. Environ Health Perspect 105:1344-1353 (1997).
83. Fenske RA, Kissel JC, Lu C, Simcox NJ, Knutson D, Koch D. Biologically based analysis of organophosphorus pesticide exposure in children in agricultural communities. Epidemiology 10(suppl):S110 (1999).
84. Nunez C, Klitzman S, Goodman A. Lead exposure among automobile radiator repair workers and their children in New York City. Am J Ind Med 23:763-777 (1993).
85. Czachur M, Stanbury M, Gerwel B, Gochfeld M, Rhoads G, Wartenberg D. A pilot study of take-home lead exposure in New Jersey. Am J Ind Med 28:289-293 (1995).
86. Schneider J, Straif K, Woitowitz H. Pleural mesothelioma and household asbestos exposure. Rev Environ Health 11:65-70 (1996).
87. Anderson H, Lilis R, Daum S, Selikoff I. Asbestosis among household contacts of asbestos factory workers. Ann N Y Acad Sci 330:387-399 (1979).
88. Piacitelli G, Whelan E, Sieber W, Gerwel B. Elevated lead contamination in homes of construction workers. Am Ind Hyg Assoc J 58:447-454 (1997).
89. Rinehart R, Yanagisawa Y. Paraoccupational exposures to lead and tin carried by electric-cable splicers. Am Ind Hyg Assoc J 54:593-599 (1993).
90. Juchau M, Faustman-Watts E. Pharmacokinetic considerations in the maternal-placental-fetal unit. Clin Obstet Gynecol 26:379-390 (1983).
91. Cresteil T. Onset of xenobiotic metabolism in children: toxicological implications. Food Addit Contam 15:45-51 (1998).
92. Renwick AG. Toxicokinetics in infants and children in relation to the ADI and TDI. Food Addit Contam 15:17-35 (1998).
93. Children's Environmental Health Network. Conference Report. In: National Research Conference on Children's Environmental Health: Research, Practice, Prevention, Policy, February 21-23, 1997, Washington, DC. Emoryville, CA:Children's Environmental Health Network/Public Health Institute, 1997.
94. Faustman E, Lewandowski T, Ponce R, Bartell S. Biologically based dose-response models for developmental toxicants: lessons from methylmercury. Inhal Toxicol 11:101-114 (1999).
95. Leroux BG, Leisenring WM, Moolgavkar SH, Faustman EM. A biologically-based dose-response model for developmental toxicology. Risk Anal 16:449-458 (1996).
Last Updated: February 16, 2000







