|
Adrenal Diseases
The Facts

|
 |
Adrenal Diseases - Congenital Adrenal Hyperplasia ( CAH )
The Facts You Need To Know
On This Page:
CAH FACT SHEET (pdf)
What is CAH?
Classical CAH-21-hydroxylase deficiency
Nonclassical CAH-21-hydroxylase deficiency
Diagnosis of CAH
Standard CAH Treatment
Newer Treatment Modalities
Prenatal Therapy
Present and Future Directions
Quick Facts About CAH
 What is Congenital Adrenal Hyperplasia?
What is Congenital Adrenal Hyperplasia?
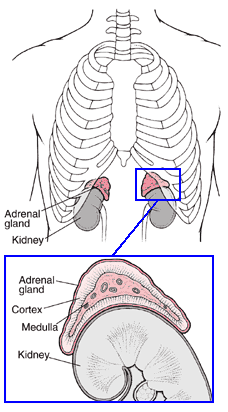 Congenital adrenal hyperplasia (CAH), also termed adrenogenital
syndrome in older literature, is a common inherited form of adrenal
insufficiency. This group of diseases is due to mutations (genetic
defects) in the genes coding for several enzymes needed to produce vital
adrenal cortex hormones. Congenital adrenal hyperplasia (CAH), also termed adrenogenital
syndrome in older literature, is a common inherited form of adrenal
insufficiency. This group of diseases is due to mutations (genetic
defects) in the genes coding for several enzymes needed to produce vital
adrenal cortex hormones.
About 95% of cases of CAH are caused because of
lack of the enzyme 21-hydroxylase. When this enzyme is missing or at
functioning at low levels, the body cannot make adequate amounts of two
vital adrenal steroid hormones: cortisol and aldosterone.
This causes
disruption in the delicate balance of hormones. Sensing low levels of cortisol, the adrenal, directed by the master hypothalamus and pituitary
glands, goes into high gear. Because cortisol production is impeded, the
adrenal cortex instead manufactures androgens, or male steroid hormones,
an undesired by-product.
In short, while one part of the adrenal
functions poorly, making inadequate amounts of cortisol and aldosterone,
another portion of the gland over-produces androgens. This last feature
distinguishes CAH-21-hydroxylase deficiency from another form of adrenal
insufficiency, Addison’s disease, since in Addisonian patients, the
adrenals are most often completely non-functional.

Classical CAH-21-hydroxylase deficiency
Lack of both cortisol and aldosterone predispose 3/4 of severely affected
individuals with CAH to "adrenal crises" with dehydration and shock, or even
death, if not properly diagnosed and treated. Excess adrenal androgen
production begins in early fetal life in classical CAH-21 affected infants,
and causes abnormal growth of the clitoris in girls and masculinization of
other genital-urinary structures. Severely affected girls may be mistaken
for boys at birth. Affected boys have no genital malformations at birth, but
continued androgen excess causes unusually fast body growth.
Inappropriately
early puberty leads to premature completion of growth and short adult
height. Proper medical treatment with the class of medications called glucocorticoids resets the abnormal balance of hormones, permits near-normal
growth and puberty. Another type of medication, mineralocorticoids, preserve
salt balance and help prevent adrenal crisis.
Proper surgical treatment by an experienced pediatric urologist
reconstructs near-normal female genitals. Some surgeons are now able to
reconstruct the vagina at the same time as they reduce the size of the
clitoris in early infancy, whereas in the past surgery was at least a
two-step process, finished in late adolescence. Some families may opt to
defer genital surgery.
Nonclassical CAH-21-hydroxylase deficiency
A milder, non-life-threatening form of CAH becomes manifest in later
childhood or even young adult life, and is not characterized by ambiguous
genitalia in girls. Rather, these individuals have a partial enzyme
deficiency, and thus have better cortisol production, normal aldosterone
production, and lower levels of adrenal androgens. They do not suffer
"adrenal crisis."
Generally, such individuals seek medical attention because
of premature development of pubic hair, irregular menstrual periods, hirsutism (unwanted body hair), or severe acne. About 10-15% of these young
women may suffer from fertility problems. Some people affected with
nonclassical CAH are not at all symptomatic, and are identified only because
of an affected relative.
Nonclassical CAH is among the most common genetic disorders, with
Ashkenazi Jews having the highest prevalence. In the general population,
depending on the ethnic breakdown of a given community, 1-5% may be affected
with non-classical CAH. Nonclassical CAH does not progress to classical CAH
in affected individuals.
The symptoms of non-classical CAH are treatable with very low dose
glucocorticoids. This type of treatment may be optional and need not be
lifelong.
Diagnosis of CAH
The diagnosis of CAH has traditionally rested on hormone measurements
combined with clinical evaluation, including history and physical
examination. Most states in the U.S. as well as several foreign countries
now perform a hormonal test for CAH within the first few days of life. These
heel-prick blood specimens are obtained at the time when blood is drawn for
thyroid tests and a number of other inherited diseases.
The rationale for
newborn screening is that mainly in boys, who have no outward sign of the
disease, the mortality from "adrenal crisis" is high, and this could be
entirely prevented by early diagnosis and medical treatment. Since the
incidence of classical CAH worldwide is about 1 in 5,000 male births (or 1
in 15,000 total births), this amounts to a substantial number of potentially
preventable infant deaths. These screening programs have achieved their
goals. Diagnostic methods are continually being refined, both for the
hormonal methods, and for the newer genetic typing discussed below.
Since the advent of molecular genetic technology, we can now examine the
genes of CAH patients and family members. This type of study has application
for prenatal testing, neonatal screening, and genetic counseling, as well as
confirming diagnosis in questionable cases. Molecular diagnosis is available
in several specialized laboratories. Families should receive genetic
counseling in conjunction with genetic testing, if they choose this
procedure.
Just as there are potential inaccuracies in hormonal testing,
there are pitfalls in genetic testing. In most cases, except in prenatal
diagnosis, people affected with classical and nonclassical forms of CAH can
be detected with hormone measurements alone, without genetic testing.
Standard CAH Treatment
Currently, standard medical treatment consists of
giving a glucocorticoid (a cortisol-like steroid medication, e.g., oral
hydrocortisone in children, or prednisone or dexamethasone in older
patients). In addition, those classical patients who have aldosterone
deficiency ("salt-wasters") need another drug, fludrocortisone (Florinef,
which acts like the missing hormone, aldosterone) to be able to retain salt.
Infants also receive supplemental salt (as crushed tablets or solutions),
whereas older patients with classical forms of CAH eat salty foods.
Although many patients are well-managed on these types of medical
regimens, it is very difficult to precisely mimic the native adrenal hormone
rhythms and achieve perfect hormonal balance. Thus, most CAH patients have
intermittent periods of fluctuating control with peaks and valleys in the
hormones doctors use to monitor the effectiveness of treatment
(specifically, 17-hydroxyprogesterone and androgens). This leads to
increases in the steroid medication doses, and sometimes these become
excessive. A known complication of high dose glucocorticoids is growth
inhibition.
Individuals with classical CAH are about 1 to 2 standard deviations below
the adult population average in height, meaning they are "short normal." A
particularly important factor in determining final height in CAH patients is
the amount of steroids given as treatment in the first 2 years of life. To
preserve height potential, children with CAH should be seen frequently by a
pediatric endocrinologist who not only measures blood hormone levels, but
also carefully assesses height, weight, blood pressure, and an annual x-ray
of the wrist (bone age x-ray).
Nonclassical CAH patients, if they require medical therapy, are usually
effectively treated with low dose hydrocortisone (children), prednisone, or
dexamethasone (the latter 2 drugs should mainly be used in older adolescents
or adults). Excessive dosing with these medications may inhibit growth in
young children, and may cause weight gain and/or hypertension. Girls with nonclassical CAH do not require genital surgery.
Newer Treatment Modalities
Because of these difficulties in fine-tuning medical treatment of
classical, severe CAH with standard therapy, some research centers have
designed experimental types of drug therapy.
One such example consists of a
4 drug combination, with an androgen blocking agent (flutamide), an
inhibitor of aromatase, an enzyme responsible for estrogen formation from
androgens (testolactone), low dose hydrocortisone and fludrocortisone.
Preliminary results after 2 years in a small group of patients are
encouraging with respect to more age-appropriate growth and less rapid bone
fusion in the experimental group. A longer trial is in progress.
Other
experimental therapies involve the use of growth hormone with or without
depot leuprolide (Lupron) to delay puberty.
It will take many years and many
more patients in clinical trials to fully understand the safety and
effectiveness of experimental therapies, since a large number of patients
will have to reach final height to determine whether the short term benefits
are sustained.
A more radical suggestion for alternative CAH therapy is a surgical one:
adrenalectomy. This therapy was in common use in the days before physicians
had access to steroid medications. It is now suggested again for selected
patients, particularly females with little- to- no enzyme activity and
severe virilization that cannot readily be controlled with medications.
Adrenalectomy can help avoid high dose glucocorticoids needed to control
persistently high adrenal androgens.
A major motivation for considering adrenalectomy is that it can now be accomplished by laparoscopy. Laparoscopy
is surgery done through one or more 1-inch incisions, with insertion of a
fiber-optic light containing a tube with openings for surgical instruments.
Laparascopic appendectomy, for instance, has minimal morbidity and low
potential for operative complications. Obviously, removing both adrenals
leaves the patient in a vulnerable Addisonian state, and one would still
have to supplement both cortisol and aldosterone equivalents.
Advocates of adrenalectomy point out that replacement hormone doses in Addisonian
patients are lower than in CAH patients, and Addisonian children do not
suffer from short stature, overweight, masculinization and ill-timed
puberty. However, if the patient is unwilling or unable to take his or her
medications, it should be understood that there are potential dire,
life-threatening consequences.
Prenatal therapy
Prenatal therapy for CAH has been practiced since 1984. It is still
considered somewhat experimental, as dexamethasone is not approved by the US
Food & Drug Administration, or by the European regulatory agencies, for this
use. In families where one child already has CAH, parents can benefit from
genetic counseling explaining how the disease is inherited, and what their
options are during subsequent pregnancies.
The aim of giving dexamethasone
to the pregnant woman at risk for a second CAH-affected child is to reduce
secretion of androgens from the female fetus’ adrenal gland, and thus reduce
the chance that the baby will be born with male-like genitals. Because
adrenal production of androgen begins in the mid-to-late first trimester
before prenatal diagnosis is done, the treatment is begun before it is known
whether the fetus is male or female, and before it is known whether the
child has CAH.
Since CAH is a recessive disease, one has a 50% chance of
inheriting a mutant gene from each carrier parent. The risk of an affected
child is thus 25% (or 50% multiplied by 50%) in each pregnancy. Since only
half of the children are female, only 1 in 8 fetuses may benefit from
prenatal treatment. Thus, 7 of 8 fetuses would be exposed unnecessarily to
steroid treatment via placental passage of the drug given to their mothers.
Several hundred children have undergone such prenatal treatment, and cursory
surveys show no major ill effects. An expert international panel urges
caution in the use of prenatal dexamethasone therapy and strict monitoring
of its application by hospital institutional review boards and ethics
committees. It is always prudent to consider the long-term potential for
unrecognized complications when experimental therapies are used.
Present and future Directions
For the present, most patients with CAH can be reasonably well-managed
with the standard diagnostic and therapeutic approaches. Molecular diagnosis
does not directly add to patient well-being, but is of use in prenatal
diagnosis and other genetic counseling.
Looking toward the future, important
diagnostic issues are demonstrating the cost-effectiveness of newer methods
of newborn screening, including molecular genetic testing. Treatment
questions to be resolved will include whether either the newer experimental
drug therapies or adrenalectomy improve patient outcome substantially, or
whether enzyme replacement by gene therapy is a possible research
breakthrough. Much work also remains in assessing outcomes among young women
with respect to psychosexual development with and without surgical
intervention.
Quick Facts About CAH
- CAH is an inherited disorder that affects the adrenal gland. In its
classical form, this disease appears in approximately 1 in 15,000 births.
- CAH is caused by a deficiency of an enzyme (adrenal steroid
21-hydroxylase) necessary for the synthesis of two vital hormones, cortisol
and aldosterone, by the human body. In its severest form, classical CAH
results in the uncontrolled loss of salt and fluids from the body, a
condition which, if undetected, can lead to adrenal crisis and death.
- One must inherit a defective enzyme trait from each parent to become
affected with CAH. This is termed an autosomal recessive disease. CAH
affects males and females in equal numbers.
- For parents who have had an affected child with CAH, there is a 25% (1 in
4) chance of producing a second affected child. Prenatal diagnosis, and
prenatal treatment of a potentially affected fetus, are available.
- Classical CAH can be detected through newborn screening. Newborn
screening for CAH saves lives.
- CAH is treatable with medications. In its classical form, CAH requires
lifelong medical management.
- Classical, severe CAH can cause genital anomalies in affected females,
with baby girls occasionally misidentified as boys. Whether and when to
consider genital surgery for females with CAH is a question to be decided on
an individual basis in conjunction with experienced health professionals.
- The non-classical form of CAH (also known as late-onset or mild CAH)
presents with milder symptoms, which may appear at any time from infancy
through adulthood.
- Non-classical CAH is not life-threatening, but can present serious
quality of life issues for the individual affected.
- The non-classical form of CAH can result in rapid growth and premature
puberty in early childhood but in some cases shorter than expected height,
hirsutism (excessive hair growth), irregular menstrual periods, acne, and
more rarely, infertility in either males or females. In young women, these
features may be confused with Apolycystic ovary syndrome.
- The symptoms of non-classical CAH are treatable with a type of steroid
hormone, glucocorticoids, in very low doses. This type of treatment may be
optional and need not be lifelong.
- Non-classical CAH is among the most common genetic disorders, with
Ashkenazi Jews having the highest prevalence. In the general population
depending on the ethnic breakdown of a given community, 1-5% may be affected
with non-classical CAH.
- Non-classical CAH does not progress to classical CAH in affected
individuals.
- Since marriage between two individuals with non-classical CAH may, in a
minority of cases, result in the birth of a child with classical CAH, those
affected by any form of CAH, or with a close family member affected by the
condition, should consider undergoing genetic counseling.
- Doctors, legislators and members of those populations most often affected
by CAH, as well as the general public, need to be educated about the disease
and its symptoms, and newborn screening for CAH will eventually become
mandatory.
In-depth information can be obtained from:
CARES Foundation, Inc.
Congenital Adrenal Hyperplasia support
Millburn, New Jersey
www.caresfoundation.org
Written by: Phyllis W. Speiser, MD
Chief, Pediatric
Endocrinology and Metabolism Division, North Shore University Hospital
Associate Professor of Pediatrics, New York University Medical College
|
