This article is based on a presentation at the Workshop on Linking Environmental Agents and Autoimmune Diseases held 1-3 September 1998 in Research Triangle Park, North Carolina.
Address correspondence to M.R. Kraine, 635 Mary Ellen Jones Bldg CB#7290, Dept. of Microbiology and Immunology, UNC, Chapel Hill, NC 27599-7290. Telephone: (919) 966-7020. Fax: (919) 962-8103. E-mail: krainemr@med.unc.edu
Received 20 January 1999; accepted 28 April 1999.
Insulin-dependent diabetes mellitus (IDDM) is an autoimmune disease characterized by monocytic and lymphocytic infiltration of the pancreatic islets. This insulitis results in the gradual destruction of the insulin-secreting ß cells found in the islets and an inability to maintain blood glucose homeostasis. As a result, diabetic patients must control blood glucose levels via exogenous insulin administration and are continually at risk for secondary complications due to fluctuations in blood glucose. Sequelae such as retinopathy, kidney failure, neuropathy, and strokes decrease the diabetic patient's quality of life and can eventually lead to premature death.
Risk for developing IDDM is strongly correlated with the expression of both major histocompatibility complex (MHC) and a number of non-MHC-related genes. However, studies of disease incidence in identical twins reveal only a 30-50% concordance rate (1). This observation has been interpreted to mean that environmental factors also influence the disease process. A number of other findings also support this notion. For example, individuals who immigrate from a country with low IDDM incidence to one with high incidence adopt the higher disease rate of the new country (2). Furthermore, northern countries tend to have higher frequencies of IDDM than do southern countries (3), and the incidence of IDDM is increasing in countries such as Sardinia that have relatively stable genetic backgrounds (4). There also appears to be an inverse relationship between the development of IDDM and socioeconomic development (5). Finally, in animal models of IDDM, diet and infection modulate disease susceptibility.
In light of these observations, investigators have for years been attempting to identify environmental factors that contribute to the development of IDDM in humans. Various synthetic drugs, diets, and infectious agents have been implicated in the inhibition and/or progression of IDDM. Unfortunately, prospective studies following these factors are rare and difficult to perform. Also, it is important to differentiate between cases of acute-onset childhood IDDM and juvenile IDDM. Both require the administration of exogenous insulin, but the mechanisms of islet destruction differ significantly between acute-onset childhood and juvenile IDDM. During acute-onset IDDM, insulin-secreting ß cells are destroyed due to direct toxicity of an agent. ß-Cell destruction occurs quite rapidly and may or may not involve an autoimmune component. In contrast, juvenile-onset IDDM is the result of monocytic- and lymphocytic-mediated ß-cell destruction. This process reflects a chronic inflammatory response during which pancreatic islets are first infiltrated with macrophages, dendritic cells, and T and B lymphocytes, and ß cells are progressively destroyed. Based on observations made in rodent models of spontaneous IDDM, T lymphocytes are the primary mediators of ß-cell destruction. Although both types of IDDM require the administration of insulin for treatment, it is the juvenile diabetes that is commonly referred to as type I IDDM.
This review summarizes the vast amount of work involved in investigating the role of environmental factors in the development of IDDM. Potential mechanisms of induction of IDDM by these factors will be reviewed. Both acute-onset and autoimmune juvenile IDDM will be covered, and the role of environmental factors in the development of each will be addressed. Studies performed in animal models, as well as studies and observations from human patients, will be included and delineated.
Molecular Mimicry
Molecular mimicry is based on two concepts involving pathogenic antigen and reactive lymphocytes (
6). The first concept is based on the observation that some pathogen proteins share sequence or structural homology with self-proteins. For example, the rubella virus capsid protein shares homology with a 52-kD pancreatic islet antigen (
7), and the coxsackie virus P2-C protein shares homology with another pancreatic antigen, glutamic acid decarboxylase (GAD) (
8,
9). The second concept is based on the fact that not all self-reactive T (and B) lymphocytes are deleted from the repertoire. They may
a) persist because of low affinity for antigen,
b) exist at a very low frequency, or
c) react with antigens that are normally sequestered in tissues. In healthy individuals, these autoreactive T lymphocytes normally exist and are not pathogenic. It is hypothesized that upon infection, pathogenic epitopes that share homology with self-epitopes are able to activate both pathogen-specific as well as self-specific T lymphocytes. Although self-specific T lymphocytes may bind epitopes with low affinity, the threshold for activation may be lowered in the context of an immune response to the pathogen. Thus, self-reactive T lymphocytes are activated and may destroy ß cells expressing the mimicked antigen.
Bystander Activation
In contrast to molecular mimicry, initial tissue injury via bystander activation is not directly antigen specific (10). For example, an infectious agent may induce cell death, resulting in the release of self-antigens that have previously been sequestered. Consequently, these antigens may activate existing autoreactive T lymphocytes. In addition, the immune response elicited by a bacterial or viral infection typically attracts antigen-presenting cells and disrupts the cytokine balance [favoring a predominantly T-helper (Th)1 environment (see "Immunoregulation")] at the site of infection/inflammation. Therefore, the body's natural response to pathogenic agents could indirectly provide an environment conducive to the activation of autoreactive T lymphocytes and development of autoimmune disease (see "Immunoregulation").
Direct Cellular Injury
Pathogens that infect pancreatic islet cells may induce IDDM by directly destroying the insulin-secreting ß cells. A critical mass of ß cells may be destroyed after repeated infections or after a single severe infection resulting in clinical IDDM.
Superantigens
Superantigens are bacterial or viral products that activate T lymphocytes expressing specific ß-chain variable gene (Vß) families of the T-cell receptor (TCR) (11). Superantigens bind the side, not the cleft, of MHC molecules, and interact with the Vß portion of the TCR. Therefore, systemic or tissue-specific infections may result in superantigen-induced activation of self-reactive T lymphocytes found in the periphery.
Immunoregulation
IDDM is an autoimmune disease in which pancreatic islet cells are infiltrated and destroyed by autoreactive CD4+ and CD8+ T lymphocytes. CD4+ Th lymphocytes (and CD8+ lymphocytes) are divided into subsets based on the type of cytokines secreted and the type of immune response mediated (12). A number of studies have demonstrated that Th1 cells are the primary mediators of ß-cell destruction leading to disease (13). Th1 cells promote inflammatory responses and are characterized by the production of interferon (IFN)-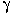 and interleukin (IL)-2. In contrast, Th2 cells confer protection from IDDM in animal models. Th2 cells promote humoral immune responses and secrete IL-4 and IL-10. Whereas IFN- and IL-12 promote the development of Th1 cells, IL-4 is necessary for differentiation of Th2 cells. Furthermore, cytokines IFN- (secreted by Th1 cells) and IL-4 and IL-10 (from Th2 cells) downregulate Th2 and Th1 effector cell differentiation and function, respectively. Thus, the Th cell and cytokine response elicited by an environmental agent may be a factor contributing to initiation and/or progression of IDDM.
and interleukin (IL)-2. In contrast, Th2 cells confer protection from IDDM in animal models. Th2 cells promote humoral immune responses and secrete IL-4 and IL-10. Whereas IFN- and IL-12 promote the development of Th1 cells, IL-4 is necessary for differentiation of Th2 cells. Furthermore, cytokines IFN- (secreted by Th1 cells) and IL-4 and IL-10 (from Th2 cells) downregulate Th2 and Th1 effector cell differentiation and function, respectively. Thus, the Th cell and cytokine response elicited by an environmental agent may be a factor contributing to initiation and/or progression of IDDM.
Drugs/Chemical Agents
Animal studies. Numerous animal models of drug-induced IDDM exist. Most of these drug regimens mediate acute-onset IDDM, making them especially useful models to study novel modalities of treatment for established disease. Streptozotocin (STZ) is an antibiotic and chemotherapeutic agent commonly used to induce IDDM in rodents. A single high dose of STZ initiates ß-cell necrosis by 4 hr, with blood glucose levels peaking (and remaining high) at 1-2 days (
14). The mechanism of high-dose STZ-induced acute-onset IDDM appears to be through damage to the mitochondria of ß cells (
15). When STZ is administered in multiple low doses, ß-cell necrosis begins by day 5 after the final injection, with blood glucose levels peaking at 1 week or later (after the final injection). This treatment regime is thought to induce a cell-mediated immune response, as ß-cell destruction continues after the STZ has been cleared from the bloodstream (
14). Therefore, the diabetes induced with multiple doses of STZ may more closely resemble the autoimmune process associated with juvenile-onset IDDM.
A second drug, alloxan, is used to induce IDDM in animal models. Alloxan is toxic to ß cells for two reasons (16). First, it rapidly accumulates in islet cells. Second, upon oxidation, it produces reactive oxygen species and peroxides that are toxic to ß cells. Consequently, the acute-onset IDDM induced by alloxan is primarily due to the accumulation of toxic byproducts in ß cells.
Cyclophosphamide is the third drug commonly used to induce IDDM in animal models. Two injections of 200 mg/kg at 14-day intervals will induce IDDM in most mice within 2-3 weeks (17,18). ß-Cell destruction is thought to be due to the elimination of regulatory T lymphocytes with concomitant upregulation of Th1 responses (17,18). mRNA expression of IFN- (a Th1 cytokine) and nitric oxide synthase is also enhanced in islet-infiltrating lymphocytes when cyclophosphamide is used to accelerate IDDM induction in the nonobese diabetic (NOD) mouse model (19).
Human studies. Though there are numerous reports of drug-induced IDDM in human patients, each case must be examined to determine its support of a drug or chemical agent as a true environmental factor influencing the disease process. For example, Vacor [1-(4-nitrophenyl)-3-(3-pyridylmethyl) urea] is probably the clearest example of a chemical that induces IDDM (20). It is a rodenticide that specifically destroys ß cells through the inhibition of mitochondrial complex I function (21). Although Vacor clearly is toxic to ß cells, it is not considered to be an important environmental factor contributing to disease incidence, as it is not normally ingested and has no clinical benefit to humans. It was, in fact, removed from the market after numerous accidental poisonings and suicide attempts in the late 1970s.
IDDM may also result from side effects of a drug administered long term, as in cases of posttransplantation immunosuppression or the treatment of cancer. In these cases, patients tend to be extremely ill and are often administered multiple medications that may interact with one another. As a result, it is difficult to determine whether the IDDM that may develop is a result of a) the underlying disease state, b) a single medication or a combination of medications, and/or c) the medication(s) involved. One such case that has recently received much attention is the use of tacrolimus (FK 506 or Prograf), a macrolide antibiotic often used as part of an immunosuppressive drug regimen. The diabetogenic effect of tacrolimus depends on numerous factors, including dose (22,23), patient age (24-26), and the dose of corticosteroids administered as part of the regimen (24). Corticosteroids alone have also been implicated as diabetogenic (because of alterations in glucose metabolism), requiring long-term insulin administration in a small number of cases (27,28). Further complicating the situation, some patients regain insulin independence when tacrolimus and/or corticosteroid doses are reduced or discontinued (24-26), whereas others do not. This also appears to be dose dependent. The mechanism by which tacrolimus causes IDDM has not been fully elucidated, but possibilities include reduced insulin mRNA synthesis and inhibition of insulin release from pancreatic ß cells, and an increase in peripheral insulin resistance (29,30). Other chronic medications that fit this category include chemotherapeutic agents such as l-asparaginase (31,32) and IFN-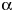 (33). Although implicated in the development of IDDM, no direct evidence for the involvement of these latter drugs or for the potential mechanism of action has been reported.
(33). Although implicated in the development of IDDM, no direct evidence for the involvement of these latter drugs or for the potential mechanism of action has been reported.
Another common occurrence is that a single drug is implicated in the development of IDDM but only in a small number of cases. For example, pentamidine treatment for Pneumocystis carinii pneumonitis has led to the induction of IDDM following ß-cell destruction (34). Although the involvement of the drug is clear, the low incidence of IDDM and the specific patient population involved also indicate that pentamidine is not a common cause of IDDM. Finally, there are occasional reports of various drugs such as isotretinoin (35) causing IDDM. It is important to note, however, that it is not known whether in these cases the drug had a direct role in the initiation or progression of the disease or if disease onset were simply coincidental with drug administration.
All cases of drug- or chemical-induced IDDM in humans to date appear to be due to rapid destruction of pancreatic ß cells and are classified as acute-onset IDDM. Furthermore, comparison of the structure and known mechanism of action of these drugs/chemicals does not reveal any unifying features that may in turn correlate with disease progression.
Food/Diet
Animal studies. Although the role of diet has been studied in animal models for more than 15 years, data on the influence of food constituents remain incomplete. A recent review by Akerblom and Knip (36) thoroughly covers the controversial role of diet and food products in the development of IDDM. Studies on the effect of diet in the BB rat have shown that plant-based diets are the most diabetogenic, possibly due to wheat and soy products (37). The ß cells of diabetic mice fed such diets exhibit increased expression of MHC I molecules and infiltration of Th cells of the Th1 phenotype (37). Cow's milk proteins, N-nitroso compounds, and gluten are other constituents that may affect the incidence of IDDM in animal models. However, some food components such as casein promote disease in the NOD mouse model but have no deleterious effect in the BB rat. As both rodent strains provide excellent models of spontaneous autoimmune IDDM, such contradictions make determination of the true role of dietary factors in disease difficult.
Human studies. Studying the effect of diet in humans is far more difficult than in animal models for the obvious reason that the human diet is usually not controlled for extended periods of time, let alone from birth. Studies of the effect of cow's milk, human breast milk, N-nitroso compounds, gluten, bovine serum albumin, fat, and protein on the development of IDDM are numerous, but none of these factors proven to play a direct role in the disease process.
Viral and Bacterial Infections
Animal studies. The role of viruses in the development of IDDM has been and continues to be a topic of great interest. The majority of animal studies involving infectious agents have been based upon induction of disease following challenge with a given virus. These studies do not necessarily reflect the development of spontaneous IDDM but nevertheless may provide insight into mechanisms by which viral infection can mediate IDDM. In addition, viral-induced models may more closely reflect acute-onset IDDM and not autoimmune-mediated juvenile-onset disease. Furthermore, spontaneous viral and bacterial infections in the NOD mouse model are known to protect mice from developing the disease. Therefore, the effect of infectious agents in diabetic animal models is still controversial.
Coxsackie-B4 (CVB) (38,39) and encephalomyocarditis M (40) are two viruses that have been used to induce IDDM in rodents. Studies in CD-1 mice challenged with a diabetogenic strain of CVB have shown that lymphocytes can destroy acutely infected islet cells, and macrophages are able to lyse chronically infected islets. This results in prolonged hyperglycemia, which reverts to normoglycemia within 6-12 months in most cases (38). Destruction of islets by macrophages can occur in the absence of detectable viral protein expression (although viral RNA is detectable), suggesting that the development of IDDM is related to prolonged presence of viral RNA in the pancreas. In an earlier study, it was observed that a) CVB infection is mouse strain specific, b) a ß-cell tropic virus is essential, and c) in most cases IDDM is transient (39). Studies using the encephalomyocarditis virus to induce IDDM are also mouse strain specific and result in a self-limiting disease.
Several groups have developed transgenic mouse models using genes encoding viral antigens and subsequent infection with the corresponding virus in an attempt to induce IDDM [reviewed in von Herrath et al. (41)]. In these models, expression of the viral antigen gene is localized to ß cells via the rat insulin promoter. Studies of the immune response after challenge with virus have provided information as to the contribution of T-lymphocyte precursor frequency, thymic negative selection, local antigen concentration, and cytokine milieu in the development of IDDM. All the mice develop IDDM because of an autoimmune process mediated by T lymphocytes, similar to the human juvenile onset disease. Despite the usefulness of these transgenic mouse models, they do not provide evidence for a direct role for viruses in the normal development of IDDM.
The role of bacterial infections in the development of IDDM has been studied through observation of disease incidence in NOD mouse colonies (incidence decreases with bacterial infection) and via administration of bacterial products in adjuvants or immunizations. Administration of complete Freund's adjuvant (CFA) does not prevent insulitis but does prevent overt IDDM in NOD mice. Animals protected from disease have decreased mRNA expression of Th1-type cytokines (42,43), increased anti-GAD67 antibodies (GAD is an autoantigen known to be important in the development of IDDM), and Th2 cytokine production (44). It is thought that Mycobacterium tuberculosis found in CFA is responsible for the protective effect of the adjuvant via immune deviation from a predominantly Th1 to a Th2 cytokine response.
A second example of protection from IDDM that implicates a bacterial product involves the bacille Calmette-Guerin (BCG) vaccine, which contains Mycobacterium bovis. BCG protects against IDDM when administered within 3 days after cyclophosphamide treatment (before the onset of insulitis in this model) (45). BCG is also thought to protect via induction of immune deviation; protected NOD mice exhibit restored anti-GAD67 antibody production and increased IL-4:IFN- ratios (45).
Human studies. In human patients, numerous viruses have a role in the development of IDDM. The mumps virus, cytomegalovirus, Epstein Barr virus, rubella virus, coxsackie viruses, and retroviruses have all been found in patients at the time of diagnosis of IDDM [reviewed in von Herrath et al. (41)]. However, it is difficult to determine if a viral infection is coincidental with the onset of IDDM or has actively contributed to the disease process that may have been ongoing for several years prior to any clinical manifestation. Therefore, the precise role viruses play (if any) in the development of IDDM is not known. For example, viruses may initiate the destruction of ß cells or promote progression of the diabetogenic response. To gain insight into these issues, it is necessary to follow a sufficient number of genetically susceptible individuals to determine whether a correlation between IDDM diagnosis and viral infections can be established from birth (and possibly in utero). Currently, this type of prospective study is extremely difficult to perform. Retrospective studies of viral infections, although easier to perform, are typically unreliable. Many infections may be subclinical and thus not recorded, and some viruses have the ability to persist in the body for several years. Together, these factors make an assessment of the role of viruses in the development of IDDM highly problematic.
Rubella, coxsackie, and retroviruses have been studied extensively in recent years. To date, rubella is the only virus for which conclusive evidence exists showing that infection significantly increases the risk of developing IDDM. In a study of 50 patients with congenital rubella, 20% of patients developed IDDM between 18 months and 33 years of age (46). Studies in which rabbits were infected in utero confirmed histologic changes in pancreatic ß cells due to the rubella virus. It is unknown whether ß-cell destruction is due to direct damage by the virus, or if the virus induces immunologic changes that indirectly destroy the islet cells. The fact that patients as young as 18 months develop IDDM would suggest a direct cytopathic effect, whereas some patients develop the disease in a more progressive manner (remaining insulin-independent until 33 years of age), suggesting indirect, possibly autoimmune, mechanisms of damage. Finally, despite an established effect on the development of IDDM, rubella virus nonetheless does not appear to be a major environmental factor influencing disease incidence.
CVB is an enterovirus that has been implicated in the development of IDDM, based on a number of observations [reviewed in Hoyt et al. (47)]. CVB infects pancreatic ß cells in vitro and in vivo and has been detected in ß cells after acute and fatal cases of IDDM. Also, IgG and IgM antibodies specific for CVB and CVB-specific RNA have been found in the sera of newly diagnosed diabetics. Seasonal variation of CVB infection has in some regions paralleled incidence of IDDM. A prospective study found relatively more frequent enteroviral infections during the first trimester of pregnancy in mothers whose children developed IDDM before 3 years of age, with no difference in children developing the disease between 4 and 6 years of age. In the same study, the group of diabetic siblings had a significantly greater number of enterovirus infections than did nondiabetic siblings (48).
Molecular mimicry, based on homology between the 2C protein of CVB and GAD, has been proposed as one mechanism by which CVB may induce IDDM. Recently, however, Horwitz et al. (49) used three mouse models (NOD, B10.H2g7, BDC2.5) to elucidate the mechanism by which CVB may induce IDDM. No spontaneous immune response to CVB cross-reactive GAD peptides was observed in NOD or B10.H2g7 mice infected with CVB, which in turn did not develop IDDM. Both these strains carry the diabetes-associated MHC class II allele I-Ag7, which presents GAD peptides to autoreactive CD4+ T lymphocytes. If CVB induces IDDM via molecular mimicry, one would expect enhanced anti-GAD T-cell reactivity and subsequent development of IDDM in infected mice. BDC2.5 mice that are transgenic for a TCR specific for an unknown pancreatic autoantigen (the majority of the T lymphocytes are specific for this ß-cell autoantigen) exhibit an enhanced onset of IDDM following infection with CVB. These results suggest that the CVB infection may induce IDDM via a bystander mechanism.
Recently, retroviral superantigens have been implicated in the development of IDDM. Pancreatic enrichment of the Vß7 family was observed in two IDDM patients, suggesting superantigen involvement (50). The superantigen implicated in those two cases was encoded by an endogenous retrovirus related to mouse mammary tumor viruses (51). The authors propose a two-step model in which systemic polyclonal activation of Vß7-restricted T cells is triggered by expression of the superantigen in the context of MHC class II. Subsequently, autoreactive T cells of the activated subset may initiate organ-specific destruction of the pancreas. Though the endogenous retrovirus has been identified, the mechanism of tissue destruction and role in disease progression as the initial trigger, final precipitator, or simply as a marker of disease remains unclear.
Although environmental factors have been implicated in the development of IDDM for years, a unifying influence remains elusive. Because of the polygenic nature of IDDM, such unifying factors may not exist. Specific environmental factors may influence individuals quite differently depending upon the genotype of that individual. Designing prospective studies that take into account genetic background of individuals also remains an obstacle to obtaining accurate data on the role of environmental agents. If these agents do influence the development of IDDM, it must be determined whether they function as initiators of disease, promote progression of an ongoing diabetogenic response, or play multiple roles in the disease process. Clearly, direct identification of environmental factors and their precise role in the disease process will be extremely useful in prevention and treatment of IDDM.
REFERENCES AND NOTES
1. Kaprio J, Tuomilehto J, Koskenvuo M, Romanov K, Reunanen A, Eriksson J, Stengard J, Kesaniemi YA. Concordance for type 1 (insulin-dependent) and type 2 (non-insulin-dependent) diabetes mellitus in a population-based cohort of twins in Finland. Diabetologia 35:1060-1067 (1992).
2. Patrick SL, Moy CS, Laporte RE. The world of insulin-dependent diabetes mellitus: what international epidemiologic studies reveal about the etiology and natural history of IDDM. Diabetes Metab Rev 5:571-578 (1989).
3. Diabetes Epidemiology Research International Group. Geographic patterns of childhood insulin-dependent diabetes mellitus. Diabetes 37:1113-1119 (1988).
4. Songini M, Muntoni S. High incidence of type-I diabetes in Sardinia [Letter]. Lancet 337:1047 (1991).
5. Singh B, Prange S, Jevnikar AM. Protective and destructive effects of microbial infection in insulin-dependent diabetes mellitus. Sem Immunol 10:79-86 (1998).
6. Oldstone MBA. Molecular mimicry and autoimmune disease. Cell 50:819-820 (1987).
7. Karounos DG, Wolinsky JS, Thomas JW. Monoclonal antibody to rubella virus capsid protein recognizes a ß-cell antigen. J Immunol 150:3080-3085 (1993).
8. Atkinson MA, Bowman MA, Campbell L, Darrow BL, Kaufman DL, Maclaren NK. Cellular immunity to a determinant common to glutamate decarboxylase and coxsackie virus in insulin-dependent diabetes. J Clin Invest 94:2125-2129 (1994).
9. Schloot NC, Roep BO, Wegmann DR, Yu L, Wang TB, Eisenbarth GS. T-cell reactivity to GAD65 peptide sequences, shared with coxsackie virus protein in recent-onset IDDM, post-onset IDDM patients and control subjects. Diabetologia 40:332-338 (1997).
10. Benoist C, Mathis D. The pathogen connection. Nature 394:227-228 (1998).
11. Luppi P, Trucco M. Superantigens in insulin-dependent diabetes mellitus. Springer Semin Immunopathol 17:333-362 (1996).
12. Mosmann TR, Coffman RL. TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol 7:145-173 (1989).
13. Katz JD, Benoist C, Mathis D. T helper cell subsets in insulin-dependent diabetes. Science 268: 1185-1190 (1995).
14. Like AA, Rossini AA. Streptozotocin-induced pancreatic insulitis: new model of diabetes mellitus. Science 193:415-417 (1976).
15. Welsh N, Paabo S, Welsh M. Decreased mitochondrial gene expression in isolated islets of rats injected neonatally with streptozotocin. Diabetologia 34:626-631 (1991).
16. Malaisse WJ. Alloxan toxicity to the pancreatic ß-cell. Biochem Pharmacol 31(22):3527-3534 (1982).
17. Harada M, Makino S. Promotion of spontaneous diabetes in non-obese diabetes-prone mice by cyclophosphamide. Diabetologia 27:604-606 (1984).
18. Yasunami R, Bach J-F. Anti-suppressor effect of cyclophosphamide on the development of spontaneous diabetes in NOD mice. Eur J Immunol 18:481-484 (1988).
19. Rothe H, Faust A, Schade U, Kleeman R, Bosse G, Hibino T, Martin S, Kolb H. Cyclophosphamide treatment of female non-obese diabetic mice causes enhanced expression of inducible nitric synthase and interferon-, but not of interleukin-4. Diabetologia 37:1154-1158 (1994).
20. Karam JH, Lewitt PA, Toung CW, Nowlain RE, Frankel BJ, Fujiya H, Freedman ZR, Grodsky GM. Insulinopenic diabetes after rodenticide (Vacor) ingestion: a unique model of acquired diabetes in man. Diabetes 31:40-45 (1980).
21. Esposti MD, Ngo A, Myers MA. Inhibition of mitochondrial complex I may account for IDDM induced by intoxication with the rodenticide Vacor. Diabetes 45:1531-1534 (1996).
22. Wagner K, Webber SA, Kurland G, Boyle GJ, Miller SA, Cipriani L, Griffith BP, Fricker FJ. New-onset diabetes mellitus in pediatric thoracic organ recipients receiving tacrolimus-based immunosuppression. J Heart Lung Transplant 16:275-282 (1996).
23. Alessiani M, Cillo U, Fung JJ, Irish W, Abu-Elmagd K, Jain A, Takaya S, Van Thiel S, Starzl TE. Adverse effects of FK 506 overdosage after liver transplantation. Transplant Proc 25:628-634 (1993).
24. Moxey-Mims M, Kay C, Light JA, Kher KK. Increased incidence of insulin-dependent diabetes mellitus in pediatric renal transplant patients receiving tacrolimus (FK506). Transplantation 65(5):617-619 (1998).
25. Fung J, Alessiani M, Abu-Elmagc K, Todo S, Shapiro R, Tzakis A, Van Thiel D, Armitage J, Jain A, McCauley J, et al. Adverse effects associated with the use of FK506. Transplant Proc 23:3105-3108 (1991).
26. Carroll PB, Rilo H, Reyes J, Alejandro R, Zeng Y, Ricordi C, Tzakis A, Shapiro R, Starzl TE. FK506 associated diabetes mellitus in the pediatric transplant population is a rare complication. Transplant Proc 23:3171-3172 (1991).
27. Perlman K, Ehrlich RM. Steroid diabetes in childhood. Am J Dis Child 136:64-68 (1982).
28. Owen OE, Cahill GF. Metabolic effects of exogenous glucocorticoids in fasted man. J Clin Invest 52:2596-2605 (1973).
29. Herold KC, Nagamatsu S, Buse JB, Kulsakdinun P, Steiner DF. Inhibition of glucose-stimulated insulin release from TC3 cells and rodent islets by an analog of FK 506. Transplantation 55:186-192 (1993).
30. Yale JF, Chamelian M, Courchesne S, Vigeant C. Peripheral insulin resistance and decreased insulin secretion after cyclosporine A treatment. Transplant Proc 20(suppl 3):985-988 (1988).
31. Gailani S, Nussbaum A, Onuma T, Freeman A. Diabetes in patients treated with asparaginase. Clin Pharmacol Ther 12:487-490 (1971).
32. Land VJ, Sutow WW, Fernbach DJ, Lane DM, Williams TE. Toxicity of l-asparaginase in children with advanced leukemia. Cancer 30:339-347 (1972).
33. Waguri M, Hanafusa T, Itoh N, Imagawa A, Miyagawa J, Kawata S, Kono N, Kuwajima M, Matsuzawa Y. Occurrence of IDDM during interferon therapy for chronic viral hepatitis. Diabetes Res Clin Prac 23:33-36 (1994).
34. Bouchard PH, Sai P, Reach G, Caubarrere I, Ganeval D, Assan R. Diabetes mellitus following pentamidine-induced hypoglycemia in humans. Diabetes 31:40-45 (1982).
35. Timperley AC, Withnall RD, Rainford DJ. The development of insulin-dependent diabetes mellitus in renal transplant patient receiving oral isotretinoin [Letter]. Nephrol Dialysis Transplant 11:753 (1996).
36. Akerblom HK, Knip M. Putative environmental factors in type I diabetes. Diabetes Metab Rev 14:31-67 (1998).
37. Scott FW. Food-induced type-I diabetes in the BB rat. Diabetes Metab Rev 12:341-359 (1996).
38. See DM, Tilles JG. Pathogenesis of virus-induced diabetes in mice. J Infect Dis 171:1131-1138 (1995).
39. Yoon J-W, Onodera T, Notkins AL. Virus-induced diabetes mellitus--ß cell damage and insulin-dependent hyperglycemia in mice infected with coxsackie virus B4. J Exp Med 148:1068-1080 (1978).
40. Notkins AL. Virus-induced diabetes mellitus--brief review. Arch Virol 54:1-17 (1977).
41. von Herrath MG, Holz A, Homann D, Oldstone MBA. Role of viruses in type I diabetes. Sem Immunol 10:87-100 (1998).
42. Shehadeh NN, La Rosa F, Lafferfty KJ. Altered cytokine activity in adjuvant inhibition of autoimmune diabetes. J Autoimmunity 6:291-296 (1991).
43. Rabinovitch A, Suarez-Pinzon WL, Sorensen O, Bleackley RC, Power RF. IFN- gene expression in pancreatic islet-infiltrating mononuclear cells correlates with autoimmune diabetes in nonobese diabetic mice. J Immunol 154:4874-4882 (1995).
44. Qin H-Y, Elliot J-F, Rajotte RV, Singh B. Endogenous immune responses to glutamic acid decarboxylase (GAD67) in NOD mice are modulated by adjuvant immunotherapy [Abstract]. Autoimmunity 24(suppl 1):A133 (1996).
45. Qin H-Y, Singh B. BCG vaccination prevents insulin-dependent diabetes mellitus (IDDM) in NOD mice after disease acceleration with cyclophosphamide. J Autoimmunity 10:271-278 (1997).
46. Menser MA, Forrest JM, Bransby RD. Rubella infection and diabetes mellitus. Lancet 1:57-60 (1978).
47. Hyoty H, Hiltunen M., Lonnrot M. Enterovirus infections and insulin-dependent diabetes mellitus--evidence for causality. Clin Diag Virol 9:77-84 (1998).
48. Hyoty H, Hiltunen M, Knip M, Laakkonen M, Vahasalo P, Karjalainen J, Koskela P, Roivainen M, Leinikki P, Hovi T et al. Childhood Diabetes in Finland (DiMe) Study Group. A prospective study of the role of coxsackie B and other enterovirus infections in the pathogenesis of IDDM. Diabetes 44:652-657 (1995).
49. Horwitz MS, Bradley LM, Harbertson J, Krahl T, Lee J, Sarvetnick N. Diabetes induced by coxsackie virus: initiation by bystander damage and not molecular mimicry. Nat Med 4(7):781-785 (1998).
50. Conrad B, Weidmann E, Trucco G, Rudert WA, Behboo R, Ricordi C, Rodriquez-Rilo H, Finegold D, Trucco M. Evidence for superantigen involvement in insulin-dependent diabetes mellitus aetiology. Nature 371:351-355 (1994).
51. Conrad B, Weissmahr RN, Boni J, Arcari R, Schupbach J, Mach B. A human endogenous retroviral superantigen as candidate autoimmune gene in type I diabetes. Cell 90:303-313 (1997).
Last Updated: September 22, 1999





