
|
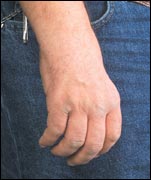
Los muchos diferentes tipos de CMT se distinguen por la edad en que
aparecen, el patrón hereditario, la severidad y si están
conectados con defectos en los axones o en la mielina. Aunque estas distinciones son útiles, es importante darse cuenta
que, debido al vasto número de defectos genéticos que
pueden llevar a CMT, algunas personas caen en los límites entre
diferentes tipos de CMT, y muchas tienen "subtipos" específicos
que no se detallan aquí. (Para mayor información sobre genética y herencia en
la CMT, ver "Es Hereditaria?",). CMT1 y CMT2Inicio: usualmente en la niñez o adolescencia Herencia: dominante autosomal Características: Estas son las dos formas más
comunes de CMT. (De hecho, un subtipo de CMT1 llamado CMT1A, causado
por un defecto en el gene PMP22 en el cromosoma 17, es responsable
de cerca de un 60 por ciento de todos los casos de CMT). La CMT1 es causada por la desmielinación y la CMT2 es causada
por axonopatía, pero ambas producen los síntomas clásicos
descritos arriba. La CMT2 se asocia a veces con una condición tratable llamada síndrome de piernas inquietas, una necesidad irresistible
de mover las piernas al estar sentado o acostado. CMT3 y CMT4
|











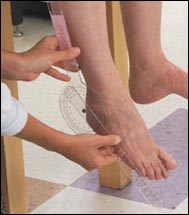

 Los médicos tienen muchas pruebas para
diagnosticar la CMT. Los médicos tienen muchas pruebas para
diagnosticar la CMT. |
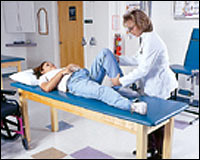
Para buscar la pérdida sensorial, el neurólogo usualmente
examinará en el paciente los reflejos de tendones profundos (como el reflejo de rodilla), los cuales se reducen o no están
presentes en la mayoría de personas con CMT.
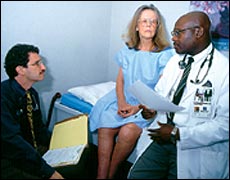
Durante esta evaluación inicial, el neurólogo también
preguntará sobre la historia familiar del paciente. Una historia
familiar con síntomas parecidos a los de la CMT, combinada con
señales de daño a los nervios en el examen físico
individual del paciente, apuntarían fuertemente hacia la CMT
u otra neuropatía hereditaria.
La ausencia de una historia familiar no descarta la CMT, pero podría
llevar al neurólogo a preguntar acerca de la diabetes, la sobre
exposición a ciertas drogas y otras causas potenciales de neuropatías.
Después, si el diagnóstico es todavía congruente
con CMT, el neurólogo podría hacer arreglos para pruebas
genéticas. Estas pruebas, hechas mediante una muestra de
sangre, están diseñadas para detectar los defectos genéticos
más comunes que se sabe que causan CMT.
Un resultado positivo de prueba genética puede llevar a un diagnóstico
definitivo y proporcionar información útil para la planificación
familiar. Pero, de nuevo, un resultado negativo no descarta la CMT.
Podría significar que el paciente tiene CMT causada por un defecto
genético desconocido, o uno tan poco comœn que no existe
una prueba para detectarlo.
El neurólogo también puede efectuar una prueba de
velocidad de conducción en los nervios (NCV), la cual mide
la fuerza y velocidad de las señales eléctricas transmitidas
a través de los nervios.
Se hace colocando electrodos de superficie, similares a los utilizados
para electrocardiogramas, sobre la piel en varios puntos encima del
nervio. Un electrodo envía un pequeño choque eléctrico
que estimula una reacción eléctrica en el nervio y los
otros registran esta respuesta a medida que viaja por el nervio. (Si
es necesario, se utiliza un anestésico local o un sedativo para
aliviar la molestia causada por los choques).
Una respuesta retrasada es una señal de desmielinación
y respuestas pequeñas son señales de axonopatía.
De esta forma, la prueba NCV se utiliza a menudo para distinguir entre
la CMT1 y la CMT2.
Otros procedimientos utilizados a veces para diagnosticar la CMT incluyen
la electromiografía (EMG), que mide las señales
eléctricas en los músculos, y con menor frecuencia, la biopsia del nervio, que involucra la remoción y el examen
de una pequeña muestra de nervio.
¿ES HEREDITARIA?
Cuando se les informa que tienen un trastorno genético como
la CMT, los pacientes a menudo preguntan, "Pero no existe en nuestra
familia, así que ¿cómo puede ser genética?"
La CMT s' puede ser hereditaria, aun cuando no exista una historia familiar
obvia. En parte, esto se debe a que la CMT puede ser heredada de tres
maneras diferentes.
X-ligada significa que el defecto genético (o mutación)
está localizado en el cromosoma X. En las mujeres, que tienen
dos cromosomas X, una copia normal del gene en un cromosoma puede a
menudo compensar (por lo menos parcialmente) la copia defectuosa. Por
lo tanto, las enfermedades ligadas a X generalmente afectan a los hombres
más severamente que a las mujeres, porque los hombres tienen
solamente un cromosoma X. Las enfermedades X-ligadas (como la CMTX)
no pueden ser pasadas de un padre a un hijo varón.
Autosomal significa que la mutación ocurre en un cromosoma que
no es X ni Y. Por lo tanto, las enfermedades autosomales afectan por
igual a hombres y mujeres. Autosomal recesiva significa que se
necesitan dos copias de un gene defectuoso para producir la enfermedad
completa. Se hereda una copia de cada progenitor, ninguno de los cuales
normalmente tendría la enfermedad. Autosomal dominante significa que una sola copia de un gene defectuoso es suficiente para
causar la enfermedad. En ese caso, una persona que hereda el gene defectuoso
de un progenitor tendrá la enfermedad, como la tendrá
también el progenitor.
Cuando la CMT se transmite en un patrón autosomal dominante,
puede ser identificada fácilmente en el árbol genealógico.
En contraste, los tipos de CMT vinculados a X o autosomal recesivos
pueden aparentar que ocurren "de la nada". Pero en realidad,
la madre o ambos progenitores pueden ser portadores que llevan en silencio
una mutación genética. Muchos padres no tienen idea que
son portadores de una enfermedad hasta que tienen un hijo con
la enfermedad.
La CMT puede de hecho ocurrir "de la nada" cuando una nueva
mutación ocurre durante la concepción del niño.
Estas son llamadas mutaciones espontáneas y una vez ocurren,
pueden ser transmitidas a la generación siguiente.
Su riesgo de heredar o transmitir la CMT depende en gran medida de
qué tipo de CMT tiene usted. Hable con el doctor o con un consejero
genético en la clínica MDA. Además, puede ver el
panfleto de la MDA "La Genética y las Enfermedades Neuromusculares".
LA BUSQUEDA DE TRATAMIENTOS Y CURAS DE LA MDA
| El sitio Web de la MDA is constantemente actualizado con la información más reciente sobre las enfermedades neuromusculares en su programa. Lea las más recientes noticias de investigación en inglés o español. |
En 1991, las causas genéticas de la CMT eran completamente
desconocidas. Pero tan sólo 10 años después, científicos
financiados por la MDA habían ayudado a identificar 10 genes
vinculados a CMT y encontrado evidencia de varios más. Este logro
ha llevado a las pruebas genéticas para muchos tipos de
CMT, lo que ha mejorado grandemente el diagnóstico y ha proporcionado
a las personas con la enfermedad mayor información para planificación
familiar.
De igual importancia, la búsqueda continua de genes de CMT ha
permitido intuir los tratamientos que podrían utilizarse
para detener o revertir el trastorno. A medida que la búsqueda
de genes de CMT se acerca a su culminación, científicos
financiados por la MDA están comenzando a investigar cómo
y por qué mutaciones genéticas específicas llevan
a diferentes tipos de CMT. En el futuro, estos conocimientos podrían
permitir a los médicos predecir con más exactitud el curso
de la CMT en pacientes individuales.
Además de los avances en genética, científicos
financiados por la MDA han hecho avances significativos en el entendimiento
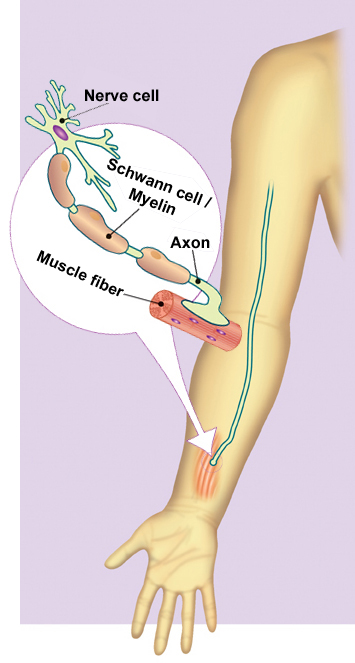
de la biología de axones y células Schwann —
las células que producen la mielina en los nervios periféricos.
La formación y el mantenimiento de la mielina parecen requerir
una interacción precisamente afinada entre los axones y las células
Schwann, y dentro de los axones existe un intrincado sistema parecido
a un ferrocarril para transportar los nutrientes de un extremo al otro.
Algunos científicos esperan poder tratar la CMT al encontrar
formas de mejorar la interacción axón-célula Schwann
o el transporte axonal.
Otros científicos están investigando la terapia genética para CMT. Con el apoyo de la MDA, un grupo está desarrollando
un método para proporcionar a los nervios dañados, genes
que codifican factores neurotróficos, proteínas que ocurren
naturalmente y que estimulan el crecimiento de las células nerviosas.
En contraste con métodos tradicionales de terapia genética
que involucran el reemplazo de un gene defectuoso con uno en funcionamiento,
este nuevo enfoque podría utilizarse para tratar todos los tipos
de CMT, sin importar el defecto base.
Finalmente, otro grupo de científicos espera tratar la CMT con células madre — células primitivas capaces de generar tipos específicos de células en el cuerpo. En experimentos de laboratorio recientes, los científicos han encontrado maneras eficientes de convertir células madre en células nerviosas y células productoras de mielina.
LA MDA ESTA AQUI PARA AYUDARLE
La Asociación de la Distrofia muscular ofrece una amplia gama
de servicios para ayudarle a usted y a su familia a tratar con la CMT
o la DS. El personal de su oficina local de la MDA está allí
para ayudarle en muchas formas. Los servicios de la Asociación
incluyen lo siguiente:
cl'nicas afiliadas a hospitales
grupos de apoyo
ayuda con la compra y
reparación de sillas de ruedas
y aparatos ortopédicos
evaluaciones de terapia física,
ocupacional y respiratoria
vacunas contra la gripe
préstamo de equipo
página electrónica en
www.mdaenespanol.org
panfletos y folletos en
español. Todos los que están
inscritos con la MDA reciben
también Quest, la revista
bimensual nacional de la MDA.
Si tuviera alguna pregunta acerca de la CMT o la DS, alguien en la MDA
le ayudará a encontrar la respuesta.
Nombre del Coordinador de los servicios de cuidado de la salud de la MDA:
Nœmero telefónico
Correo electrónico
Línea nacional de ayuda de la MDA: (800) 572-1717
ENFERMEDADES INCLUIDAS EN EL PROGRAMA DE LA MDA
Las siguientes enfermedades neuromusculares están incluidas
dentro de los
proyectos de investigación y los programas de asistencia de la
Asociación de la Distrofia Muscular:
DISTROFIAS MUSCULARES
Distrofia muscular (seudohipertrófica)
de Duchenne
Distrofia muscular de Becker
Distrofia muscular de Emery-Dreifuss
Distrofia muscular del anillo -seo
Distrofia muscular facioescapulo-
humeral (de Landouzy-Dejerine)
Distrofia miotónica
(enfermedad de Steinert)
Distrofia muscular oculofaríngea
Distrofia muscular distal
Distrofia muscular congénita
ENFERMEDADES DE LAS NEURONAS MOTORAS
Esclerosis lateral amiotrófica
Atrofia muscular espinal progresiva
infantil (Tipo 1, enfermedad de
Werdnig-Hoffmann)
Atrofia muscular espinal intermedia
(Tipo 2)
Atrofia muscular espinal juvenil
(Tipo 3, enfermedad de
Kugelberg-Welander)
Atrofia muscular espinal adulta
Atrofia muscular espinal bulbar
(enfermedad de Kennedy)
MIOPATIAS INFLAMATORIAS
Polimiositis
Dermatomiositis
Miositis de cuerpo de inclusión
ENFERMEDADES DE LA UNION NEUROMUSCULAR
Miastenia grave
S'ndrome (miasténico) de Lambert-Eaton
S'ndrome miasténico congénito
ENFERMEDADES DE LOS NERVIOS PERIFERICOS
Enfermedad de Charcot-Marie-Tooth
(atrofia muscular peroneal)
Ataxia de Friedreich
Enfermedad de Dejerine-Sottas
ENFERMEDADES METABOLICAS DEL MUSCULO
Deficiencia de fosforilasa
(enfermedad de McArdle)
Deficiencia de maltasa ac'dica
(enfermedad de Pompe)
Deficiencia de fosfofructokinasa
(enfermedad de Tarui)
Deficiencia de enzimas bifurcadoras
(enfermedad de Cori o de Forbes)
Miopatía mitocóndrica
Deficiencia de carnitina
Deficiencia de transferasa de palmitil
carnitina
Deficiencia de kinasa de fosfoglicerato
Deficiencia de mutasa de fosfoglicerato
Deficiencia de deshidrogenasa de lactato
Deficiencia de desaminasa de miodenilato
MIOPATIAS DEBIDAS A ANORMALIDADES ENDOCRINAS
Miopatía hipertiroidea
Miopatía hipotiroidea
OTRAS MIOPATIAS
Mioton'a congénita
Paramiotonía congénita
Enfermedad del núcleo central
Miopatía nemalina
Miopatía miotubular
Parálisis periódica
ASOCIACION DE LA DISTROFIA MUSCULAR
Combatiendo enfermedades neuromusculares ¥ 3300 East Sunrise Drive,
Tucson, AZ 85718
(800) 572-1717 • www.mda.org, www.mdaenespanol.org
Jerry Lewis, Líder Nacional
© 2002, Muscular Dystrophy Association
![]()
| Facts About Charcot-Marie-Tooth Disease and Dejerine-Sottas Disease
|
|
