|
|
Reviewed November 2006
What is mitochondrial DNA?
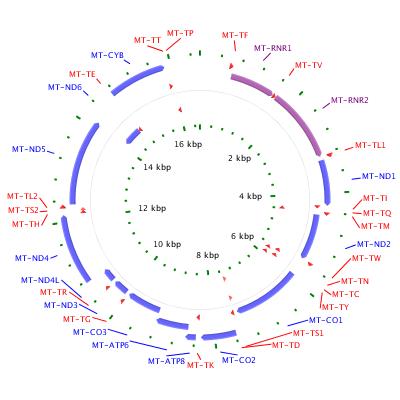
Mitochondria are structures within cells that convert the energy from food into a form that cells can use. Although most DNA is packaged in chromosomes within the nucleus, mitochondria also have a small amount of their own DNA. This genetic material is known as mitochondrial DNA or mtDNA. In humans, mitochondrial DNA spans about 16,500 DNA building blocks (base pairs), representing a fraction of the total DNA in cells.
Mitochondrial DNA contains 37 genes, all of which are essential for normal mitochondrial function. Thirteen of these genes provide instructions for making enzymes involved in oxidative phosphorylation. Oxidative phosphorylation is a process that uses oxygen and simple sugars to create adenosine triphosphate (ATP), the cell's main energy source. The remaining genes provide instructions for making molecules called transfer RNAs (tRNAs) and ribosomal RNAs (rRNAs), which are chemical cousins of DNA. These types of RNA help assemble protein building blocks (amino acids) into functioning proteins.
Mitochondrial genes are among the estimated 20,000 to 25,000 total genes in the human genome.
There are genetic conditions related to mitochondrial genes.
What conditions are related to mitochondrial DNA?
The following conditions are related to changes in mitochondrial DNA.
- cancers
-
Mitochondrial DNA is prone to noninherited (somatic) mutations. Somatic mutations occur in the DNA of certain cells during a person’s lifetime and typically are not passed to future generations. Somatic mutations in mitochondrial DNA have been reported in some forms of cancer, including breast, colon, stomach, liver, and kidney tumors. These mutations also have been associated with cancer of blood-forming tissue (leukemia) and cancer of immune system cells (lymphoma).
Somatic mutations in mitochondrial DNA may increase the production of potentially harmful molecules called reactive oxygen species. Mitochondrial DNA is particularly vulnerable to the effects of these molecules and has a limited ability to repair itself. As a result, reactive oxygen species easily damage mitochondrial DNA, causing a buildup of additional somatic mutations. Researchers continue to investigate how these mutations may lead to uncontrolled cell division and the growth of cancerous tumors.
-
Leber hereditary optic neuropathy
-
Mutations in four mitochondrial genes, MT-ND1, MT-ND4, MT-ND4L, and MT-ND6, have been identified in people with Leber hereditary optic neuropathy. These genes provide instructions for making proteins that are part of a large enzyme complex. This group of enzymes, known as complex I, is necessary for oxidative phosphorylation. The mutations responsible for Leber hereditary optic neuropathy change single protein building blocks (amino acids) in these proteins, which may affect the generation of ATP within mitochondria. It remains unclear, however, why the effects of these mutations are often limited to the nerve that relays visual information from the eye to the brain (the optic nerve). Additional genetic and environmental factors probably contribute to the vision loss and other medical problems associated with Leber hereditary optic neuropathy.
-
mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes
-
Mutations in at least five mitochondrial genes, MT-ND1, MT-ND5, MT-TH, MT-TL1, and MT-TV, can cause the characteristic features of mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes (MELAS). Some of these genes provide instructions for making proteins that are part of a large enzyme complex, called complex I, that is necessary for oxidative phosphorylation. The other genes provide instructions for making transfer RNA molecules, which are essential for protein production within mitochondria.
Mutations in one transfer RNA gene, MT-TL1, cause more than 80 percent of all cases of MELAS. These mutations impair the ability of mitochondria to make proteins, use oxygen, and produce energy. Researchers have not determined how changes in mitochondrial DNA lead to the specific signs and symptoms of MELAS. They continue to investigate the effects of mitochondrial gene mutations in different tissues, particularly in the brain.
-
neuropathy, ataxia, and retinitis pigmentosa
-
Mutations in one mitochondrial gene, MT-ATP6, have been found in people with neuropathy, ataxia, and retinitis pigmentosa (NARP). The MT-ATP6 gene provides instructions for making a protein that is essential for normal mitochondrial function. This protein forms one part (subunit) of an enzyme called ATP synthase. This enzyme, which is also known as complex V, is responsible for the last step of oxidative phosphorylation, in which a molecule called adenosine diphosphate (ADP) is converted to ATP. Mutations in the MT-ATP6 gene alter the structure or function of ATP synthase, reducing the ability of mitochondria to make ATP. It is unclear how this disruption in mitochondrial energy production leads to muscle weakness, vision loss, and the other specific features of NARP.
-
nonsyndromic deafness
-
Mutations in two mitochondrial genes, MT-RNR1 and MT-TS1, are associated with nonsyndromic deafness (hearing loss without related signs and symptoms affecting other parts of the body). These genes provide instructions for making types of RNA. The MT-RNR1 gene provides instructions for a specific type of ribosomal RNA called 12S RNA. A particular form of transfer RNA, designated as tRNASer(UCN), is formed from the MT-TS1 gene. Both of these RNA molecules help assemble amino acids into full-length, functioning proteins within mitochondria.
Mutations in the MT-RNR1 gene increase the risk of hearing loss, particularly in people who take antibiotic medications called aminoglycosides. These antibiotics are typically used to treat chronic bacterial infections such as tuberculosis. Aminoglycosides kill bacteria by binding to their ribosomal RNA and disrupting the bacteria's ability to make proteins. Genetic changes in the MT-RNR1 gene often make the 12S RNA in human cells look similar to bacterial ribosomal RNA. As a result, aminoglycosides can target the altered 12S RNA just as they target bacterial ribosomal RNA. The antibiotic easily binds to the abnormal 12S RNA, which impairs the ability of mitochondria to produce proteins needed for oxidative phosphorylation. Researchers believe that this unintended effect of aminoglycosides may reduce the amount of ATP produced in mitochondria, increase the production of harmful byproducts, and eventually cause the cell to self-destruct (undergo apoptosis).
Nonsyndromic deafness also results from genetic changes in the MT-TS1 gene. Most of the mutations change a single building block (nucleotide) in the tRNASer(UCN) molecule. These mutations likely disrupt the normal production of the molecule or alter its structure. As a result, less tRNASer(UCN) is available to assemble proteins within mitochondria. These changes reduce the production of proteins needed for oxidative phosphorylation, which may impair the ability of mitochondria to make ATP.
Researchers have not determined why the effects of mutations in the MT-RNR1 and MT-TS1 genes are usually limited to cells in the inner ear that are essential for hearing. They believe that other genetic or environmental factors must play a role in the signs and symptoms associated with these mutations.
- other disorders
-
Inherited changes in mitochondrial DNA can cause problems with growth, development, and function of the body's systems. These mutations disrupt the mitochondria's ability to efficiently generate energy for the cell. Conditions caused by mutations in mitochondrial DNA often involve multiple organ systems. The effects of these conditions are most pronounced in organs and tissues that require a lot of energy (such as the heart, brain, and muscles). Although the health consequences of inherited mitochondrial DNA mutations vary widely, some frequently observed features include muscle weakness and wasting, problems with movement, diabetes, kidney failure, heart disease, loss of intellectual functions (dementia), hearing loss, and abnormalities involving the eyes and vision.
A buildup of noninherited (somatic) mutations in mitochondrial DNA has been associated with an increased risk of certain age-related disorders such as heart disease, Alzheimer disease, and Parkinson disease. Additionally, research suggests that the progressive accumulation of these mutations over a person's lifetime may play a role in the normal process of aging.
Is there a standard way to diagram mitochondrial DNA?
Mitochondrial DNA is typically diagrammed as a circular structure with genes and regulatory regions labeled.
Where can I find additional information about mitochondrial DNA?
You may find the following resources about mitochondrial DNA helpful. These materials are written for the general public.
- Additional NIH Resources - National Institutes of Health
-
You may also be interested in these resources, which are designed for genetics professionals and researchers.
-
-
Gene Tests - DNA tests ordered by healthcare professionals (5 links)
-
- PubMed
 - Recent literature - Recent literature
-
OMIM - Genetic disorder catalog (2 links)
- Map Viewer - Genetic maps
Where can I find general information about mitochondria?
The Handbook provides basic information about genetics in clear language.
These links provide additional genetics resources that may be useful.
What glossary definitions help with understanding mitochondrial DNA?
The resources on this site should not be used as a substitute for
professional medical care or advice. Users seeking information about
a personal genetic disease, syndrome, or condition should consult with a qualified
healthcare professional.
See How can I find a genetics professional in my area? in the Handbook.
|