

|
|
(Posted: 09/13/2002)
How to Find a Safety-Related Labeling ChangeUse the drop-down menu to select a product by brand name. |
MedWatch
Home | What's New | About
Medwatch | How to Report | Submit
Report | Safety Info
Continuing Education | Download
PDF | Comments | Privacy
Statement
ACCUPRIL (quinapril HCl) Tablets
[July 22, 2002: Pfizer]
PRECAUTIONS
Drug Interaction
Other Agents
Co-administration of multiple 10 mg doses of atorvastatin with 80 mg of ACCUPRIL resulted in no significant change in the steady-state pharmacokinetic parameters of atorvastatin.
OVERDOSAGE
No data are available with respect to overdosage in humans.
Doses of 1440 to 4280 mg/kg of quinapril cause significant lethality in mice and rats.
No specific information is available on the treatment of overdosage with quinapril. The most likely clinical manifestation would be symptoms attributable to severe hypotension.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
[July 12, 2002: Takeda]
[Other safety related information: http://www.fda.gov/medwatch/SAFETY/2002/safety02.htm#thiazo]
[Other labeling changes not appearing in 2002 PDR: http://www.fda.gov/medwatch/SAFETY/2002/jan02.htm#actos]
PRECAUTIONS
Drug Interactions
The following drugs were studied in healthy volunteers with a co-administration of ACTOS 45 mg once daily. Listed below are the results:
Fexofenadine HCl: Co-administration of ACTOS for 7 days with 60 mg fexofenadine administered orally twice daily resulted in no significant effect on pioglitazone pharmacokinetics. ACTOS had no significant effect on fexofenadine pharmacokinetics.
Ranitidine HCl: Co-administration of ACTOS for 7 days with ranitidine administered orally twice daily for either 4 or 7 days resulted in no significant effect on pioglitazone pharmacokinetics. ACTOS showed no significant effect on ranitidine pharmacokinetics.
Nifedipine ER: Co-administration of ACTOS for 7 days with 30 mg nifedipine ER administered orally once daily for 4 days to male and female volunteers resulted in a loge transformed AUC ratio of 0.88 (CI 0.81 - 0.95). In view of the high variability of nifedipine pharmacokinetics, the clinical significance of this finding is unknown.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
AGGRASTAT (tirofiban HCl) Injection
[July 24, 2002: Merck]
[Other labeling changes not appearing in 2002 PDR: http://www.fda.gov/medwatch/SAFETY/2002/jun02.htm#aggras ]
DOSAGE AND ADMINISTRATION
Directions for Use
AGGRASTAT may be administered in the same intravenous line as atropine sulfate, dobutamine, dopamine, epinephrine HCl, furosemide, lidocaine, midazolam, HCl, morphine sulfate, nitroglycerin, potassium chloride, propranolol HCl, and PEPCID* (famotidine) Injection. AGGRASTAT should not be administered in the same intravenous line as diazepam.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
AREDIA (pamidronate disodium) Injection
[July 12, 2002: Novartis]
WARNINGS
DUE TO THE RISK OF CLINICALLY SIGNIFICANT DETERIORATION IN RENAL FUNCTION, WHICH MAY PROGRESS TO RENAL FAILURE, SINGLE DOSES OF AREDIA SHOULD NOT EXCEED 90 MG (See DOSAGE AND
ADMINISTRATION for appropriate infusion durations). Bisphosphonates, including Aredia, have been associated with renal toxicity manifested as deterioration of renal function and potential renal failure. Patients who receive Aredia should have serum creatinine assessed prior to each treatment.
Patients treated with Aredia for bone metastases should have the dose withheld if renal function has deteriorated. (See DOSAGE AND ADMINISTRATION).
Patients who receive an intravenous infusion of Aredia should have periodic evaluations of standard laboratory and clinical parameters of renal function.
PREGNANCY:
AREDIA SHOULD NOT BE USED DURING PREGNANCY. Aredia may cause fetal harm when administered to a pregnant woman, (See PRECAUTIONS, Pregnancy Category D). There are no studies in pregnant women using Aredia. If the patient becomes pregnant while taking this drug, the patient should be apprised of the potential harm to the fetus. Women of childbearing potential should be advised to avoid becoming pregnant.
PRECAUTIONS
General
Renal Insufficiency: Aredia is excreted intact primarily via the kidney, and the risk of renal adverse reactions may be greater in patients with impaired renal function. Patients who receive Aredia should have serum creatinine assessed prior to each treatment. In patients receiving Aredia for bone metastases, who show evidence of deterioration in renal function, Aredia treatment should be withheld until renal function returns to baseline (See WARNINGS and DOSAGE AND ADMINISTRATION.)
For the treatment of bone metastases, the use of Aredia in patients with severe renal impairment is not recommended.
Drug Interactions
Caution is indicated when Aredia is used with other potentially nephrotoxic drugs.
Pregnancy
Pregnancy Category changed from "C" to "D," and (See Warnings).
ADVERSE REACTIONS
Clinical Studies
Osteolytic Bone Metastases of Breast Cancer and Osteolytic Lesions of Multiple Myeloma
Renal Toxicity
In a study of the safety and efficacy of Aredia 90 mg (2 hour infusion) versus Zometa 4 mg (15 minute infusion) in bone metastases patients with multiple myeloma of breast cancer, renal deterioration was defined as an increase in serum creatinine of 0.5 mg/dL for patients with normal baseline creatinine (<1.4 mg/dL) or an increase of 1.0 mg/dL for patients with an abnormal baseline creatinine ³ 1.4 mg/dL). The following are data on the incidence of renal deterioration in patients in this trial. See Table below.
Incidence of Renal Function Deterioration in Multiple Myeloma and Breast Cancer
Patients with Normal and Abnormal Serum Creatinine at Baseline*
|
Patient Population/Baseline Creatinine |
Aredia 90 mg/2 hours |
Zometa 4 mg/15 minutes |
|
n/N (%) |
n/N (%) |
|
|
Normal |
20/246 (8.1%) |
23/246 (9.3%) |
|
Abnormal |
2/22 (9.1%) |
1/26 (3.8%) |
|
Total |
22/268 (8.2%) |
24/272 (8.8%) |
*Patients were randomized following the 15-minute infusion amendment for the Zometa arm.
OVERDOSAGE
Single doses of Aredia should not exceed 90 mg and the duration of the intravenous infusion should be no less than 2 hours. (See WARNINGS.)
DOSAGE AND ADMINISTRATION
Osteolytic Bone Lesions of Multiple Myeloma &
Osteolytic Bone Metastases of Breast Cancer
Patients who receive Aredia should have serum creatinine assessed prior to each treatment. Treatment should be withheld for renal deterioration. In a clinical study, renal deterioration was defined as follows:
In this clinical study, Aredia treatment was resumed only when the creatinine returned to within 10% of the baseline value.
Preparation of Solution
Reconstitution
Method of Administration DUE TO THE RISK OF CLINICALLY SIGNIFICANT DETERIORATION IN RENAL FUNCTION, WHICH MAY PROGRESS TO RENAL FAILURE, SINGLE DOSES OF AREDIA SHOULD NOT EXCEED 90 MG. (SEE WARNINGS).
There must be strict adherence to the intravenous administration recommendations for Aredia in order to decrease the risk of deterioration in renal function.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
AZULFIDINE (sulfasalazine) Tablets and EN-tabs
[July 30, 2002: Pharmacia & Upjohn]
Labeling provides for revision to the DESCRIPTION, CLINICAL PHARMACOLOGY, INDICATIONS AND USAGE, CONTRAINDICATIONS, WARNINGS, PRECAUTIONS, ADVERSE REACTIONS, OVERDOSAGE AND DOSAGE AND ADMINISTRATION sections for the AZULFIDINE Tablet package insert for the 100-count and 300-count packages in order to harmonize the AZULFIDINE Tablet insert with the AZULFIDINE EN tabs insert.
Contact the company for a copy of the labeling/package insert.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
CLINIMIX (amino acid in dextrose) &
CLINIMIX E (amino acid in dextrose with electrolytes) Injection
[July 25, 2002: Baxter Healthcare]
WARNINGS:
Added at end of WARNINGS section:
WARNING: This product contains aluminum that may be toxic.
Aluminum may reach toxic levels with prolonged parenteral administration if kidney function is impaired. Premature neonates are particularly at risk because their kidneys are immature, and they require large amounts of calcium and phosphate solutions, which contain aluminum.
Research indicates that patients with impaired kidney function, including premature neonates, who receive parenteral levels of aluminum at greater than 4 to 5 m g/kg/day accumulate aluminum at levels associated with central nervous system and bone toxicity. Tissue loading may occur at even lower rates of administration.
PRECAUTIONS:
Added at end of PRECAUTIONS section:
Drug product contains no more than 25 m g/L of aluminum.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
[July 5, 2002: Novartis]
[Other safety related information: http://www.fda.gov/medwatch/SAFETY/2002/safety02.htm#clozar]
[Other labeling changes not appearing in 2002 PDR: http://www.fda.gov/medwatch/SAFETY/2002/jan02.htm#cloraz]
PRECAUTIONS
Cardiomyopathy
Cases of cardiomyopathy have been reported in patients treated with clozapine. The reporting rate for cardiomyopathy in clozapine-treated patients in the United States (8.9 per 100,000 person-years) was similar to an estimate of the cardiomyopathy incidence in the United States general population derived from the 1999 National Hospital Discharge Survey data (9.7 per 100,000 person-years). Approximately 80% of clozapine-treated patients in whom cardiomyopathy was reported were less than 50 years of age; the duration of treatment with clozapine prior to cardiomyopathy diagnosis varied, but was >6 months in 65% of the reports. Dilated cardiomyopathy was most frequently reported, although a large percentage of reports did not specify the type of cardiomyopathy. Signs and symptoms suggestive of cardiomyopathy, particularly exertional dyspnea, fatigue, orthopnea, paroxysmal nocturnal dyspnea, and peripheral edema should alert the clinician to perform further investigations. If the diagnosis of cardiomyopathy is confirmed, the prescriber should discontinue clozapine unless the benefit to the patient clearly outweighs the risk.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
CORTIFOAM (hydrocortisone acetate) Aerosol
[July 8, 2002: Schwartz Pharma]
Labeling extensively revised. Contact the company for a copy of the new label/package insert.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
DHE 45 (dihydroergotamine) Injection
Migranal (dihydroergotamine)Nasal Spray
[July 31, 2002: Xcel Pharmaceuticals]
Prescribing Information
|
WARNING Serious and/or life-threatening peripheral ischemia has been associated with the coadministration of DIHYDROERGOTAMINE with potent CYP 3A4 inhibitors including protease inhibitors and macrolide antibiotics. Because CYP 3A4 inhibition elevates the serum levels of DIHYDROERGOTAMINE, the risk for vasospasm leading to cerebral ischemia and/or ischemia of the extremities is increased. Hence, concomitant use of these medications is contraindicated. (See also CONTRAINDICATIONS and WARNINGS section) |
CLINICAL PHARMACOLOGY
Pharmacokinetics:
Interactions
Pharmcokinetic interactions (increased blood levels) have been reported in patients treated orally with dihydroergotamine and macrolide antibiotics, primarily troleandomycin, presumably due to inhibition of cytochrome P450 3A metabolism of dihydroergotamine by troleandomycin. Dihydroergotamine has also been shown to be an inhibitor of cytochrome P450 3A catalyzed reactions. No pharmacokinetic interactions involving other cytochrome P450 isoenzymes are known.
Replaced by:
Pharmacokinetic interactions have been reported in patients treated orally with other ergot alkaloids (e.g., increased levels of ergotamine) and macrolide antibiotics, principally troleandomycin, presumably due to inhibition of cytochrome P450 3A metabolism of the alkaloids by troleandomycin. Dihydroergotamine has also been shown to be an inhibitor of cytochrome P450 3A catalyzed reactions and rare reports of ergotism have been obtained from patients treated with dihydroergotamine and macrolide antibiotics (e.g., troleandomycin, clarithromycin, erythromycin), and in patients treated with dihydroergotamine and protease inhibitors (e.g. ritonavir), presumably due to inhibition of cytochrome P450 3A metabolism of ergotamine (See CONTRAINDICATIONS). No pharmacokinetic interactions involving other cytochrome P450 isoenzymes are known.
CONTRAINDICATIONS
New first paragraph [Bolded text for Migranal label]:
There have been a few reports of serious adverse events associated with the coadministration of dihydroergotamine and potent CYP 3A4 inhibitors, such as protease inhibitors and macrolide antibiotics, resulting in vasospasm that led to cerebral ischemia and/or ischemia of the extremities. The use of potent CYP 3A4 inhibitors (ritonavir, nelfinavir, indinavir, erythromycin, clarithromycin, troleandomycin, ketoconazole, itraconazole) with dihydroergotamine is, therefore contraindicated (See WARNINGS: CYP 3A4 Inhibitors).
WARNINGS
D.H.E. 45 (dihydroergotamine mesylate) Injection, USP should only be used where a clear diagnosis of migraine headache has been established.
Migranal (dihydroergotamine mesylate, USP) Nasal Spray should only be used where a clear diagnosis of migraine headache has been established.
CYP 3A4 Inhibitors (e.g. Macrolide Antibiotics and Protease Inhibitors)
There have been rare reports of serious adverse events in connection with the coadministration of dihydroergotamine and potent CYP 3A4 inhibitors, such as protease inhibitors and macrolide antibiotics, resulting in vasospasm that led to cerebral ischemia and/or and ischemia of the extremities. The use of potent CYP 3A4 inhibitors with dihydroergotamine should therefore be avoided (see CONTRAINDICATIONS). Examples of some of the more potent CYP 3A4 inhibitors include: anti-fungals ketoconazole and itraconazole, the protease inhibitors ritonavir, nelfinavir, and indinavir, and macrolide antibiotics erythromycin, clarithromycin, and troleandomycin. Other less potent CYP 3A4 inhibitors should be administered with caution. Less potent inhibitors include saquinavir, nefazodone, fluconazole, grapefruit juice, fluoxetine, fluvoxamine, zileuton, and clotrimazole. These lists are not exhaustive, and the prescriber should consider the effects on CYP3A4 of other agents being considered for concomitant use with dihydroergotamine.
Fibrotic Complications
There have been reports of pleural and retroperitoneal fibrosis in patients following prolonged daily use of injectable dihydroergotamine mesylate. Rarely, prolonged daily use of other ergot alkaloid drugs has been associated with cardiac valvular fibrosis. Rare cases have also been reported in association with the use of injectable dihydroergotamine mesylate; however, in those cases, patients also received drugs known to be associated with cardiac valvular fibrosis.
[D.H.E.]
Administration of D.H.E. 45 (dihydroergotamine mesylate) Injection, USP, should not exceed the dosing guidelines and should not be used for chronic daily administration (see DOSAGE AND ADMINISTRATION).
[Migranal]
Administration of Migranal (dihydroergotamine mesylate, USP) Nasal Spray, should not exceed the dosing guidelines and should not be used for chronic daily administration (see DOSAGE AND ADMINISTRATION).
PRECAUTIONS
General
Last sentence
Fibrotic Complications: see WARNINGS: Fibrotic Complications
Information for Patients
New last paragraphs:
Administration of D.H.E. 45 (dihydroergotamine mesylate) Injection , USP, should not exceed the dosing guidelines and should not be used for chronic daily administration (see DOSAGE AND ADMINISTRATION).
Administration of Migranal (dihydroergotamine mesylate, USP) Nasal Spray, should not exceed the dosing guidelines and should not be used for chronic daily administration (see DOSAGE AND ADMINISTRATION).
Drug Interactions
[Between Nicotine and SSRI's]
CYP 3A4 Inhibitors (e.g. Macrolide Antibiotics and Protease Inhibitors)
See CONTRAINDICATIONS and WARNINGS.
ADVERSE REACTIONS
Last sentence of first paragraph:
[D.H.E. and Migranol]
Fibrotic complications have been reported in association with long term use of injectable dihydroergotamine mesylate (see WARNINGS: Fibrotic Complications).
DOSAGE AND ADMINISTRATION
D.H.E. 45 (dihydroergotamine mesylate) Injection, USP, should not be used for chronic daily administration.
Migranal (dihydroergotamine mesylate, USP) Nasal Spray, should not be used for chronic daily administration.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
DOBUTREX (dobutamine) Injection
[July 23, 2002: Eli Lilly]
PRECAUTIONS
Geriatric Use
Of the 1893 patients in clinical studies who were treated with dobutamine, 930 (49.1%) were 65 and older. No overall differences in safety or effectiveness were observed between these and younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or drug therapy.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
ELOCON (mometasone furoate) Cream, Lotion & Ointment
[July 17, 2002: Schering]
[In the PRECAUTIONS, Carcinogenesis, Mutagenesis, Impairment of Fertility:, Pregnancy: Teratogenic Effects: and Pediatric Use: sections the underlined, green text largely replaces previous text.]
[Cream]
CLINICAL PHARMACOLOGY
Pharmacokinetics:
Third paragraph added -
In a study evaluating the effects of mometasone furoate cream on the hypothalamic-pituitary- adrenal (HPA) axis, 15 grams were applied twice daily for 7 days to 6 adult patients with psoriasis or atopic dermatitis. The cream was applied without occlusion to at least 30% of the body surface. The results show that the drug caused a slight lowering of adrenal corticosteroid secretion.
Last paragraph added -
Ninety-seven pediatric patients ages 6 to 23 months, with atopic dermatitis, were enrolled in an open-label, hypothalamic-pituitary-adrenal (HPA) axis safety study. ELOCON Cream was applied once daily for approximately 3 weeks over a mean body surface area of 41% (range 15% to 94%). In approximately 16% of patients who showed normal adrenal function by Cortrosyn test before starting treatment, adrenal suppression was observed at the end of treatment with ELOCON Cream. The criteria for suppression were: basal cortisol level of £ 5 mcg/dL, 30- minute post-stimulation level of £ 18 mcg/dL, or an increase of <7 mcg/dL. Follow-up testing 2 to 4 weeks after stopping treatment, available for 5 of the patients, demonstrated suppressed HPA axis function in one patient, using these same criteria.
PRECAUTIONS
Information for Patients:
8. Other corticosteroid-containing products should not be used with ELOCON Cream without first consulting with the physician.
Pediatric Use:
ELOCON Cream caused HPA axis suppression in approximately 16% of pediatric
patients ages 6 to 23 months, who showed normal adrenal function by Cortrosyn test before starting treatment, and were treated for approximately 3 weeks over a mean body surface area of 41% (range 15% to 94%). The criteria for suppression were: basal cortisol level of ≤5 mcg/dL, 30-minute post-stimulation level of ≤18 mcg/dL, or an increase of <7 mcg/dL. Follow-up testing 2 to 4 weeks after study completion, available for 5 of the patients, demonstrated suppressed HPA axis function in one patient, using these same criteria. Long-term use of topical corticosteroids has not been studied in this population (see CLINICAL PHARMACOLOGY – Pharmacokinetics Section).
Carcinogenesis, Mutagenesis, Impairment of Fertility:
Long-term animal studies have not been performed to evaluate the carcinogenic potential of ELOCON (mometasone furoate cream) Cream, 0.1%. Long-term carcinogenicity studies of mometasone furoate were conducted by the inhalation route in rats and mice. In a 2-year carcinogenicity study in Sprague-Dawley rats, mometasone furoate demonstrated no statistically significant increase of tumors at inhalation doses up to 67 mcg/kg (approximately 0.04 times the estimated maximum clinical topical dose from ELOCON Cream on a mcg/m 2 basis). In a 19-month carcinogenicity study in Swiss CD-1 mice, mometasone furoate demonstrated no statistically significant increase in the incidence of tumors at inhalation doses up to 160 mcg/kg (approximately 0.05 times the estimated maximum clinical topical dose from ELOCON Cream on a mcg/m 2 basis).
Mometasone furoate increased chromosomal aberrations in an in vitro Chinese hamster ovary cell assay, but did not increase chromosomal aberrations in an in vitro Chinese hamster lung cell assay. Mometasone furoate was not mutagenic in the Ames test or mouse lymphoma assay, and was not clastogenic in an in vivo mouse micronucleus assay, a rat bone marrow chromosomal aberration assay, or a mouse male germ-cell chromosomal aberration assay. Mometasone furoate also did not induce unscheduled DNA synthesis in vivo in rat hepatocytes.
In reproductive studies in rats, impairment of fertility was not produced in male or female rats by subcutaneous doses up to 15 mcg/kg (approximately 0.01 times the estimated maximum clinical topical dose from ELOCON Cream on a mcg/m 2 basis).
Pregnancy:
Teratogenic Effects: Pregnancy Category C: Corticosteroids have been shown to be teratogenic in laboratory animals when administered systemically at relatively low dosage levels. Some corticosteroids have been shown to be teratogenic after dermal application in laboratory animals.
When administered to pregnant rats, rabbits, and mice, mometasone furoate increased fetal malformations. The doses that produced malformations also decreased fetal growth, as measured by lower fetal weights and/or delayed ossification. Mometasone furoate also caused dystocia and related complications when administered to rats during the end of pregnancy.
In mice, mometasone furoate caused cleft palate at subcutaneous doses of 60 mcg/kg and above. Fetal survival was reduced at 180 mcg/kg. No toxicity was observed at 20 mcg/kg. (Doses of 20, 60 and 180 mcg/kg in the mouse are approximately 0.01, 0.02 and 0.05 times the estimated maximum clinical topical dose from ELOCON Cream on a mcg/m 2 basis).
In rats, mometasone furoate produced umbilical hernias at topical doses of 600 mcg/kg and above. A dose of 300 mcg/kg produced delays in ossification, but no malformations. (Doses of 300 and 600 mcg/kg in the rat are approximately 0.2 and 0.4 times the estimated maximum clinical topical dose from ELOCON Cream on a mcg/m 2 basis). In rabbits, mometasone furoate caused multiple malformations (e.g., flexed front paws, gallbladder agenesis, umbilical hernia, hydrocephaly) at topical doses of 150 mcg/kg and above (approximately 0.2 times the estimated maximum clinical topical dose from ELOCON Cream on a mcg/m 2 basis). In an oral study, mometasone furoate increased resorptions and caused cleft palate and/or head malformations (hydrocephaly and domed head) at 700 mcg/kg. At 2800 mcg/kg most litters were aborted or resorbed. No toxicity was observed at 140 mcg/kg. (Doses of 140, 700 and 2800 mcg/kg in the rabbit are approximately 0.2, 0.9 and 3.6 times the estimated maximum clinical topical dose from ELOCON Cream on a mcg/m 2 basis).
When rats received subcutaneous doses of mometasone furoate throughout pregnancy or during the later stages of pregnancy, 15 mcg/kg caused prolonged and difficult labor and reduced the number of live births, birth weight and early pup survival. Similar effects were not observed at 7.5 mcg/kg. (Doses of 7.5 and 15 mcg/kg in the rat are approximately 0.005 and 0.01 times the estimated maximum clinical topical dose from ELOCON Cream on a mcg/m 2 basis).
There are no adequate and well-controlled studies of teratogenic effects from topically applied corticosteroids in pregnant women. Therefore, topical corticosteroids should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Pediatric Use:
ELOCON Cream caused HPA axis suppression in approximately 16% of pediatric patients ages 6 to 23 months, who showed normal adrenal function by Cortrosyn test before starting treatment, and were treated for approximately 3 weeks over a mean body surface area of 41% (range 15% to 94%). The criteria for suppression were: basal cortisol level of £ 5 mcg/dL, 30-minute post-stimulation level of £ 18 mcg/dL, or an increase of <7 mcg/dL. Follow-up testing 2 to 4 weeks after study completion, available for 5 of the patients, demonstrated suppressed HPA axis function in one patient, using these same criteria. Long-term use of topical corticosteroids has not been studied in this
population (see CLINICAL PHARMACOLOGY – Pharmacokinetics Section).
Geriatric Use: Clinical studies of ELOCON Cream included 190 subjects who were 65 years of age and over and 39 subjects who were 75 years of age and over. No overall differences in safety or effectiveness were observed between these subjects and younger subjects, and other reported clinical experience has not identified differences in responses between the elderly and younger patients. However, greater sensitivity of some older individuals cannot be ruled out.
ADVERSE REACTIONS
The following adverse reactions were reported to be possibly or probably related to treatment with ELOCON Cream during clinical studies in 4% of 182 pediatric patients 6 months to 2 years of age: decreased glucocorticoid levels, 2; paresthesia, 2; folliculitis, 1; moniliasis, 1; bacterial infection, 1; skin depigmentation, 1. The following signs of skin atrophy were also observed among 97 patients treated with ELOCON Cream in a clinical study: shininess 4, telangiectasia 1, loss of elasticity 4, loss of normal skin markings 4, thinness 1, and bruising 1. Striae were not observed in this study.
[Lotion]
CLINICAL PHARMACOLOGY
Like other topical corticosteroids, mometasone furoate has anti-inflammatory, anti-pruritic, and vasoconstrictive properties. The mechanism of the anti-inflammatory activity of the topical steroids, in general, is unclear. However, corticosteroids are thought to act by the induction of phospholipase A2 inhibitory proteins, collectively called lipocortins. It is postulated that these proteins control the biosynthesis of potent mediators of inflammation such as prostaglandins and leukotrienes by inhibiting the release of their common precursor arachidonic acid. Arachidonic acid is released from membrane phospholipids by phospholipase A2.
Pharmacokinetics:
[Largely replaces previous text]
The extent of percutaneous absorption of topical corticosteroids is determined by many factors including the vehicle and the integrity of the epidermal barrier and the use of occlusive dressings. (See DOSAGE AND ADMINISTRATION). Topical corticosteroids can be absorbed from normal intact skin. Occlusive dressings with hydrocortisone for up to 24 hours have not been demonstrated to increase penetration; however, occlusion of hydrocortisone for 96 hours markedly enhances penetration. Studies in humans indicate that approximately 0.7% of the applied dose of ELOCON Ointment, 0.1%, enters the circulation after 8 hours of contact on normal skin without occlusion. A similar minimal degree of absorption of the corticosteroid from the lotion formulation would be anticipated. Inflammation and/or other disease processes in the skin may increase percutaneous absorption.
Studies performed with ELOCON Lotion indicate that it is in the medium range of potency as compared with other topical corticosteroids.
In a study evaluating the effects of mometasone furoate lotion on the hypothalamic-pituitary- adrenal (HPA) axis, 15 mL were applied without occlusion twice daily (30 mL per day) for 7 days to 4 adult patients with scalp and body psoriasis. At the end of treatment, the plasma cortisol levels for each of the 4 patients remained within the normal range and changed little from baseline.
Sixty-five pediatric patients ages 6 to 23 months, with atopic dermatitis, were enrolled in an open-label, hypothalamic-pituitary-adrenal (HPA) axis safety study. ELOCON Lotion was applied once daily for approximately 3 weeks over a mean body surface area of 40% (range 16% to 90%). In approximately 29% of patients who showed normal adrenal function by Cortrosyn test before starting treatment, adrenal suppression was observed at the end of treatment with ELOCON Lotion. The criteria for suppression were: basal cortisol level of £ 5 mcg/dL, 30- minute post-stimulation level of £ 18 mcg/dL, or an increase of <7 mcg/dL. Follow-up testing 2 to 4 weeks after stopping treatment, available for 8 of the patients, demonstrated suppressed HPA axis function in one patient, using these same criteria.
INDICATIONS AND USAGE
ELOCON Lotion is indicated for the relief of the inflammatory and pruritic manifestations of corticosteroid-responsive dermatoses.
ELOCON Lotion, 0.1%, is a medium potency corticosteroid indicated for the relief of the inflammatory and pruritic manifestations of corticosteroid-responsive dermatoses.
Since safety and efficacy of ELOCON Lotion have not been established in pediatric patients below 12 years of age, its use in this age group is not recommended. (see PRECAUTIONS - Pediatric Use).
PRECAUTIONS
Information for Patients:
6. This medication should not be used on the face, underarms, or groin areas unless directed by the physician.
7. As with other corticosteroids, therapy should be discontinued when control is achieved. If no improvement is seen within 2 weeks, contact the physician.
8. Other corticosteroid-containing products should not be used with ELOCON Lotion without first consulting with the physician.
Carcinogenesis, Mutagenesis, Impairment of Fertility: Long-term animal studies have not been performed to evaluate the carcinogenic potential of ELOCON (mometasone furoate lotion) Lotion. Long-term carcinogenicity studies of mometasone furoate were conducted by the inhalation route in rats and mice. In a 2-year carcinogenicity study in Sprague-Dawley rats, mometasone furoate demonstrated no statistically significant increase of tumors at inhalation doses up to 67 mcg/kg (approximately 0.04 times the estimated maximum clinical topical dose from ELOCON Lotion on a mcg/m 2 basis). In a 19-month carcinogenicity study in Swiss CD-1 mice, mometasone furoate demonstrated no statistically significant increase in the incidence of tumors at inhalation doses up to 160 mcg/kg (approximately 0.05 times the estimated maximum clinical topical dose from ELOCON Lotion on a mcg/m 2 basis).
Mometasone furoate increased chromosomal aberrations in an in vitro Chinese hamster ovary cell assay, but did increase chromosomal aberrations in an in vitro Chinese hamster lung cell assay. Mometasone furoate was not mutagenic in the Ames test or mouse lymphoma assay, and was not clastogenic in an in vivo mouse micronucleus assay, a rat bone marrow chromosomal aberration assay, or a mouse male germ-cell chromosomal aberration assay. Mometasone furoate also did not induce unscheduled DNA synthesis in vivo in rat hepatocytes.
In reproductive studies in rats, impairment of fertility was not produced in male or female rats by subcutaneous doses up to 15 mcg/kg (approximately 0.01 times the estimated maximum clinical topical dose from ELOCON Lotion on a mcg/m 2 basis).
Genetic toxicity studies with mometasone furoate, which included the Ames test, mouse lymphoma assay, and a micronucleus test, did not reveal any mutagenic potential.
Pregnancy:
Teratogenic Effects: Pregnancy Category C:
Corticosteroids are generally teratogenic in laboratory animals when administered systemically at relatively low dosage levels. Corticosteroids have been shown to be teratogenic after dermal application in laboratory animals.
Corticosteroids have been shown to be teratogenic in laboratory animals when administered systemically at relatively low dosage levels. Some corticosteroids have been shown to be teratogenic after dermal application in laboratory animals.
When administered to pregnant rats, rabbits, and mice, mometasone furoate increased fetal malformations. The doses that produced malformations also decreased fetal growth, as measured by lower fetal weights and/or delayed ossification. Mometasone furoate also caused dystocia and related complications when administered to rats during the end of pregnancy.
In mice, mometasone furoate caused cleft palate at subcutaneous doses of 60 mcg/kg and above. Fetal survival was reduced at 180 mcg/kg. No toxicity was observed at 20 mcg/kg. (Doses of 20, 60 and 180 mcg/kg in the mouse are approximately 0.01, 0.02 and 0.05 times the estimated maximum clinical topical dose from ELOCON Lotion on a mcg/m 2 basis).
In rats, mometasone furoate produced umbilical hernias at topical doses of 600 mcg/kg and above. A dose of 300 mcg/kg produced delays in ossification, but no malformations. (Doses of 300 and 600 mcg/kg in the rat are approximately 0.2 and 0.4 times the estimated maximum clinical topical dose from ELOCON Lotion on a mcg/m2 basis).
In rabbits, mometasone furoate caused multiple malformations (e.g., flexed front paws, gallbladder agenesis, umbilical hernia, hydrocephaly) at topical doses of 150 mcg/kg and above (approximately 0.2 times the estimated maximum clinical topical dose from ELOCON Lotion on a mcg/m2 basis). In an oral study, mometasone furoate increased resorptions and caused cleft palate and/or head malformations (hydrocephaly and domed head) at 700 mcg/kg. At 2800 mcg/kg most litters were aborted or resorbed. No toxicity was observed at 140 mcg/kg. (Doses of 140, 700 and 2800 mcg/kg in the rabbit are approximately 0.2, 0.9 and 3.6 times the estimated maximum clinical topical dose from ELOCON Lotion on a mcg/m2 basis).
When rats received subcutaneous doses of mometasone furoate throughout pregnancy or during the later stages of pregnancy, 15 mcg/kg caused prolonged and difficult labor and reduced the number of live births, birth weight and early pup survival. Similar effects were not observed at 7.5 mcg/kg. (Doses of 7.5 and 15 mcg/kg in the rat are approximately 0.005 and 0.01 times the estimated maximum clinical topical dose from ELOCON Lotion on a mcg/m 2 basis).
Drugs of this class should not be used extensively on pregnant patients, in large amounts, or for prolonged periods.
There are no adequate and well-controlled studies of teratogenic effects from topically applied corticosteroids in pregnant women. Therefore, topical corticosteroids should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Nursing Mothers: It is not known whether topical administration of corticosteroids could result in sufficient systemic absorption to produce detectable quantities in human milk. Systemically administered corticosteroids are secreted into breast milk in quantities not likely to have a deleterious effect on the infant. Systemically administered corticosteroids appear in human milk and could suppress growth, interfere with endogenous corticosteroid production, or cause other untoward effects. Because many drugs are excreted in human milk, caution should be exercised when ELOCON Lotion is administered to a nursing woman. Nevertheless, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
Pediatric Use:
Previous subsection replaced with the following:
Since safety and efficacy of ELOCON Lotion have not been established in pediatric patients below 12 years of age, its use in this age group is not recommended.
ELOCON Lotion caused HPA axis suppression in approximately 29% of pediatric patients ages 6 to 23 months who showed normal adrenal function by Cortrosyn test before starting treatment, and were treated for approximately 3 weeks over a mean body surface area of 40% (range 16% to 90%). The criteria for suppression were: basal cortisol level of £ 5 mcg/dL, 30-minute post-stimulation level of £ 18 mcg/dL, or an increase of <7 mcg/dL. Follow-up testing 2 to 4 weeks after stopping treatment, available for 8 of the patients, demonstrated suppressed HPA axis function in one patient, using these same criteria. Long-term use of topical corticosteroids has not been studied in this population. (see CLINICAL PHARMACOLOGY -Pharmacokinetics).
Because of a higher ratio of skin surface area to body mass, pediatric patients are at a greater risk than adults of HPA axis suppression and Cushing's syndrome when they are treated with topical corticosteroids. They are, therefore, also at greater risk of glucocorticosteroid insufficiency during and/or after withdrawal of treatment. Pediatric patients may be more susceptible than adults to skin atrophy, including striae, when they are treated with topical corticosteroids. Pediatric patients applying topical corticosteroids to greater than 20% of body surface are at higher risk of HPA axis suppression.
HPA axis suppression, Cushing's syndrome, linear growth retardation, delayed weight gain and intracranial hypertension have been reported in pediatric patients receiving topical corticosteroids. Manifestations of adrenal suppression in children include low plasma cortisol levels and absence of response to ACTH stimulation. Manifestations of intracranial hypertension include bulging fontanelles, headaches, and bilateral papilledema.
ELOCON (mometasone furoate lotion) Lotion should not be used in the treatment of diaper dermatitis.
Geriatric Use: Clinical studies of ELOCON Lotion did not include sufficient number of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious.
ADVERSE REACTIONS
The following local adverse reactions were reported with ELOCON Lotion during clinical studies with 209 patients: acneiform reaction, 2; burning, 4; and itching, 1. In an irritation/sensitization study with 156 normal subjects, folliculitis was reported in 4.
In clinical studies involving 209 patients, the incidence of adverse reactions associated with the use of ELOCON Lotion was 3%. Reported reactions included acneiform reaction, 2; burning, 4; and itching, 1. In an irritation/sensitization study involving 156 normal subjects, the incidence of folliculitis was 3% (4 subjects).
The following adverse reactions were reported to be possibly or probably related to treatment with ELOCON Lotion during a clinical study, in 14% of 65 pediatric patients 6 months to 2 years of age: decreased glucocorticoid levels, 4; paresthesia, 2; dry mouth, 1; an unspecified endocrine disorder, 1; pruritus, 1; and an unspecified skin disorder, 1. The following signs of skin atrophy were also observed among 65 patients treated with ELOCON Lotion in a clinical study: shininess 4, telangiectasia 2, loss of elasticity 2, and loss of normal skin markings 3. Striae, thinness and bruising were not observed in this study.
The following additional local adverse reactions have been reported infrequently when other topical dermatologic corticosteroids have been used as recommended. with topical corticosteroids, but may occur more frequently with the use of occlusive dressings. These reactions are listed in an approximate decreasing order of occurrence: burning, itching, irritation, dryness, folliculitis, hypertrichosis, acneiform eruptions, hypopigmentation, perioral dermatitis, allergic contact dermatitis, maceration of the skin, secondary infection, skin atrophy, striae, and miliaria.
DOSAGE AND ADMINISTRATION
Apply a few drops of ELOCON Lotion to the affected skin areas once daily and massage lightly until it disappears. For the most effective and economical use, hold the nozzle of the bottle very close to the affected areas and gently squeeze. Since safety and efficacy of ELOCON Lotion have not been established in pediatric patients below 12 years of age, its use in this age group is not recommended. (see PRECAUTIONS - Pediatric Use).
As with other corticosteroids, therapy should be discontinued when control is achieved. If no improvement is seen within 2 weeks, reassessment of diagnosis may be necessary. ELOCON Lotion should not be used with occlusive dressings unless directed by a physician. ELOCON Lotion should not be applied in the diaper area if the patient requires diapers or plastic pants as these garments may constitute occlusive dressing.
[Ointment]
CLINICAL PHARMACOLOGY
Pharmacokinetics:
Third paragraph added -
In a study evaluating the effects of mometasone furoate ointment on the hypothalamic-pituitary-adrenal (HPA) axis, 15 grams were applied twice daily for 7 days to six adult patients with psoriasis or atopic dermatitis. The ointment was applied without occlusion to at least 30% of the body surface. The results show that the drug caused a slight lowering of adrenal corticosteroid secretion.
Last paragraph added -
Sixty-three pediatric patients ages 6 to 23 months, with atopic dermatitis, were enrolled in an open-label, hypothalamic-pituitary-adrenal (HPA) axis safety study. ELOCON Ointment was applied once daily for approximately 3 weeks over a mean body surface area of 39% (range 15% to 99%). In approximately 27% of patients who showed normal adrenal function by Cortrosyn test before starting treatment, adrenal suppression was observed at the end of treatment with ELOCON Ointment. The criteria for suppression were: basal cortisol level of £ 5 mcg/dL, 30-minute post-stimulation level of £ 18 mcg/dL, or an increase of <7 mcg/dL. Follow-up testing 2 to 4 weeks after stopping treatment, available for 8 of the patients, demonstrated suppressed HPA axis function in 3 patients, using these same criteria.
PRECAUTIONS
Information for Patients:
8. Other corticosteroid-containing products should not be used with ELOCON Ointment without first consulting with the physician.
Carcinogenesis, Mutagenesis, Impairment of Fertility:
Long-term animal studies have not been performed to evaluate the carcinogenic potential of ELOCON (mometasone furoate ointment) Ointment. Long-term carcinogenicity studies of mometasone furoate were conducted by the inhalation route in rats and mice. In a 2-year carcinogenicity study in Sprague-Dawley rats, mometasone furoate demonstrated no statistically significant increase of tumors at inhalation doses up to 67 mcg/kg (approximately 0.04 times the estimated maximum clinical topical dose from ELOCON Ointment on a mcg/m 2 basis). In a 19-month carcinogenicity study in Swiss CD-1 mice, mometasone furoate demonstrated no statistically significant increase in the incidence of tumors at inhalation doses up to 160 mcg/kg (approximately 0.05 times the estimated maximum clinical topical dose from ELOCON Ointment on a mcg/m 2 basis).
Mometasone furoate increased chromosomal aberrations in an in vitro Chinese hamster ovary cell assay, but did not increase chromosomal aberrations in an in vitro Chinese hamster lung cell assay. Mometasone furoate was not mutagenic in the Ames test or mouse lymphoma assay, and was not clastogenic in an in vivo mouse micronucleus assay, a rat bone marrow chromosomal aberration assay, or a mouse male germ-cell chromosomal aberration assay. Mometasone furoate also did not induce unscheduled DNA synthesis in vivo in rat hepatocytes.
In reproductive studies in rats, impairment of fertility was not produced in male or female rats by subcutaneous doses up to 15 mcg/kg (approximately 0.01 times the estimated maximum clinical topical dose from ELOCON Ointment on a mcg/m 2 basis).
Pregnancy:
Teratogenic Effects: Pregnancy Category C:
Corticosteroids have been shown to be teratogenic in laboratory animals when administered systemically at relatively low dosage levels. Some corticosteroids have been shown to be teratogenic after dermal application in laboratory animals.
When administered to pregnant rats, rabbits, and mice, mometasone furoate increased fetal malformations. The doses that produced malformations also decreased fetal growth, as measured by lower fetal weights and/or delayed ossification. Mometasone furoate also caused dystocia and related complications when administered to rats during the end of pregnancy.
In mice, mometasone furoate caused cleft palate at subcutaneous doses of 60 mcg/kg and above. Fetal survival was reduced at 180 mcg/kg. No toxicity was observed at 20 mcg/kg. (Doses of 20, 60 and 180 mcg/kg in the mouse are approximately 0.01, 0.02 and 0.05 times the estimated maximum clinical topical dose from ELOCON Ointment on a mcg/m 2 basis).
In rats, mometasone furoate produced umbilical hernias at topical doses of 600 mcg/kg and above. A dose of 300 mcg/kg produced delays in ossification, but no malformations. (Doses of 300 and 600 mcg/kg in the rat are approximately 0.2 and 0.4 times the estimated maximum clinical topical dose from ELOCON Ointment on a mcg/m 2 basis). In rabbits, mometasone furoate caused multiple malformations (e.g., flexed front paws, gallbladder agenesis, umbilical hernia, hydrocephaly) at topical doses of 150 mcg/kg and above (approximately 0.2 times the estimated maximum clinical topical dose from ELOCON Ointment on a mcg/m 2 basis). In an oral study, mometasone furoate increased resorptions and caused cleft palate and/or head malformations (hydrocephaly and domed head) at 700 mcg/kg. At 2800 mcg/kg most litters were aborted or resorbed. No toxicity was observed at 140 mcg/kg. (Doses of 140, 700 and 2800 mcg/kg in the rabbit are approximately 0.2, 0.9 and 3.6 times the estimated maximum clinical topical dose from ELOCON Ointment on a mcg/m 2 basis).
When rats received subcutaneous doses of mometasone furoate throughout pregnancy or during the later stages of pregnancy, 15 mcg/kg caused prolonged and difficult labor and reduced the number of live births, birth weight and early pup survival. Similar effects were not observed at 7.5 mcg/kg. (Doses of 7.5 and 15 mcg/kg in the rat are approximately 0.005 and 0.01 times the estimated maximum clinical topical dose from ELOCON Ointment on a mcg/m 2 basis).
There are no adequate and well-controlled studies of teratogenic potential of mometasone furoate effects from topically applied corticosteroids in pregnant women. Therefore, topical corticosteroids should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Pediatric Use:
ELOCON Ointment caused HPA axis suppression in approximately 27% of pediatric patients ages 6 to 23 months, who showed normal adrenal function by Cortrosyn test before starting treatment, and were treated for approximately 3 weeks over a mean body surface area of 39% (range 15% to 99%). The criteria for suppression were: basal cortisol level of ≤5 mcg/dL, 30-minute post-stimulation level of ≤18 mcg/dL, or an increase of <7 mcg/dL. Follow-up testing 2 to 4 weeks after stopping treatment, available for 8 of the patients, demonstrated suppressed HPA axis function in 3 patients, using these same criteria. Long-term use of topical corticosteroids has not been studied in this population. (see CLINICAL PHARMACOLOGY -Pharmacokinetics).
Geriatric Use: Clinical studies of ELOCON Ointment included 310 subjects who were 65 years of age and over and 57 subjects who were 75 years of age and over. No overall differences in safety or effectiveness were observed between these subjects and younger subjects, and other reported clinical experience has not identified differences in responses between the elderly and younger patients. However, greater sensitivity of some older individuals cannot be ruled out.
ADVERSE REACTIONS
Following paragraph added:
The following adverse reactions were reported to be possibly or probably related to treatment with ELOCON Ointment during a clinical study, in 5% of 63 pediatric patients 6 months to 2 years of age: decreased glucocorticoid levels, 1; an unspecified skin disorder, 1; and a bacterial skin infection, 1. The following signs of skin atrophy were also observed among 63 patients treated with ELOCON Ointment in a clinical study: shininess 4, telangiectasia 1, loss of elasticity 4, loss of normal skin markings 4, thinness 1. Striae and bruising were not observed in this study.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
FORTOVASE (saquinavir) Capsules
[July 2, 2002:Hoffman-La Roche]
WARNINGS:
ALERT: Find out about medicines that should not be taken with FORTOVASE. This statement is included on the product’s bottle label.
Concomitant use of FORTOVASE and St. John’s wort (hypericum perforatum) or products containing St. John’s wort is not recommended. Coadministration of protease inhibitors, including FORTOVASE, with St. John’s wort is expected to substantially decrease protease inhibitor concentrations and may result in sub-optimal levels of FORTOVASE and lead to loss of virologic response and possible resistance to FORTOVASE or to the class of protease inhibitors.
PRECAUTIONS
Information for Patients
A statement to patients and health care providers is included on the product’s bottle label: ALERT: Find out about medicines that should NOT be taken with FORTOVASE. A Patient Package Insert (PPI) for FORTOVASE is available for patient information.
FORTOVASE may interact with some drugs; therefore, patients should be advised to report to their doctor the use of any other prescription, nonprescription medication, or herbal products, particularly St. John’s wort.
Patient Information About FORTOVASE
ALERT: Find out about medicines that should NOT be taken with FORTAVASE. Please also read the section MEDICINES YOU SHOULD NOT TAKE WITH FORTOVASE.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
FOSAMAX (alendronate sodium) Tablets
[July 25, 2002: Merck]
[Labeling information not found in 2001 PDR]
WARNINGS
Second paragraph, first sentence -
Esophageal adverse experiences, such as esophagitis, esophageal ulcers and esophageal erosions, occasionally with bleeding and rarely followed by esophageal stricture or perforation, have been reported in patients receiving treatment with FOSAMAX."
ADVERSE REACTIONS
Post-Marketing Experience Gastrointestinal: esophagitis, esophageal erosions, esophageal ulcers, rarely esophageal stricture or perforation, and oropharyngeal ulceration .
[once weekly dose PPI/once daily dose PPI]
What are the possible side effects of FOSAMAX?
First sentence -
Some patients may develop severe digestive reactions including irritation, inflammation or ulceration (occasionally severe and/or with bleeding) of the esophagus (the tube that connects your mouth with your stomach).
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
[July 11, 2002: Novo Nordisk]
INDICATIONS AND USAGE:
Added to end of section:
Because GlucaGen depletes glycogen stores, the patient should be given oral carbohydrates as soon as the procedure is completed.
DOSAGE AND ADMINISTRATION:
Added to end of section:
When the diagnostic procedure is over, give oral carbohydrates to restore the liver glycogen and prevent occurrence of secondary hypoglycemia.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
[July 3, 2002: Hoffman-La Roche]
PRECAUTIONS
Pregnancy:
Antiretroviral Pregnancy Registry: To monitor maternal-fetal outcomes of pregnant women exposed to HIVID, an Antiretroviral Pregnancy Registry has been established. Physicians are encouraged to register patients by calling 1-800-258-4263.
Nursing Mothers:The Centers for Disease Control and Prevention recommend
HIV-infected mothers not breastfeed their infants to avoid risking postnatal transmission of HIV. It is not known whether zalcitabine is excreted in human milk. Because of both the potential for HIV transmission and the potential for serious adverse reactions in nursing infants, mothers should be instructed not to breastfeed if they are receiving HIVID.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
INVIRASE (saquinavir mesylate) Capsules
[July 2, 2002: Hoffman-La Roche]
[Other labeling changes not in 2002 PDR: http://www.fda.gov/medwatch/SAFETY/2002/jun02.htm#invira ]
WARNINGS
ALERT: Find out about medicines that should not be taken with INVIRASE. This statement is included on the product’s bottle label.
Concomitant use of INVIRASE and St. John’s wort (hypericum perforatum) or products containing St. John’s wort is not recommended. Coadministration of protease inhibitors, including INVIRASE, with St. John’s wort is expected to substantially decrease protease inhibitor concentrations and may result in sub-optimal levels of INVIRASE and lead to loss of virologic response and possible resistance to INVIRASE or to the class of protease inhibitors.
PRECAUTIONS: Information for Patients
A statement to patients and health care providers is included on the product’s bottle label: ALERT: Find out about medicines that should NOT be taken with INVIRASE.
INVIRASE may interact with some drugs; therefore, patients should be advised to report to their doctor the use of any other prescription, non-prescription medication, or herbal products, particularly St. John’s wort.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
ISOVUE-M (Iopamidol) Injection
[July 8, 2002: Bracco Diagnostics]
[Other labeling changes not in 2002 PDR: http://www.fda.gov/medwatch/SAFETY/2002/jun02.htm#isovue]
WARNINGS
Contrast media may promote sickling in individuals who are homozygous for sickle cell disease when injected intraveneously or intra-arterially. Although Isovue-M is not injected intravascularly, measurable plasma levels are attained after intrathecal administration of iopamidol.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
LEVOXYL (levothyroxine sodium) Tablets
[July 17, 2002: Jones Pharma]
WARNINGS
|
WARNING: Thyroid hormones, including LEVOXYL, either alone or with other therapeutic agents, should not be used for the treatment of obesity or for weight loss. In euthyroid patients, doses within the range of daily hormonal requirements are ineffective for weight reduction. Larger doses may produce serious or even life threatening manifestations of toxicity, particularly when given in association with sympathomimetic amines such as those used for their anorectic effects. |
ADVERSE REACTIONS:
Pseudotumor cerebri has been reported in children receiving levothyroxine therapy and slipped capital femoral epiphysis. Overtreatment may result in craniosynostosis in infants and premature closure of the epiphyses in children with resultant compromised adult height.
[In 2002 label]
DOSAGE AND ADMINISTRATION
Myxedema Coma --Myxedema coma is a life-threatening emergency characterized by poor circulation and hypometabolism, and may result in unpredictable absorption of levothyroxine sodium from the gastrointestinal tract. Therefore, oral levothyroxine is not recommended to treat this condition. Intravenous levothyroxine sodium should be administered.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
[July 9, 2002: Pharmacia & Upjohn]
PRECAUTIONS
Geriatric Use
Clinical studies of LONITEN Tablets did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
HOW SUPPLIED
Store at controlled room temperature 15
°
to 30 °
C (59 °
to 86 °
F) 20
°
to 25 °
C (68 °
to 77 °
F) [see USP].
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
MANDOL (cefamandole nafate) Injection
[July 18, 2002: Eli Lilly]
Labeling provides for an updated [extensively revised] Microbiology subsection of the CLINICAL PHARMACOLOGY section and revisions to WARNINGS, PRECAUTIONS, and ADVERSE REACTIONS sections. Contact the company for a copy of the new labeling/package insert.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
M.V.I. Pediatric (Multi-Vitamins) Injection
[July 8, 2002: NeoSan Pharmaceuticals]
[Other labeling changes regarding Multi-Vitamins Injection not in 2002 PDR:
http://www.fda.gov/medwatch/SAFETY/2002/jun02.htm#mvi12]
WARNINGS
WARNING:This product contains aluminum that may be toxic. Aluminum may reach toxic levels with prolonged parenteral administration if kidney function is impaired. Premature neonates are particularly at risk because their kidneys are immature, and they require large amounts of calcium and phosphate solutions, which contain aluminum.
Research indicates that patients with impaired kidney function, including premature neonates, who receive parenteral levels of aluminum at greater than 4 to 5 m g/kg/day accumulate aluminum at levels associated with central nervous system and bone toxicity. Tissue loading may occur at even lower rates of administration.
IMMEDIATE CONTAINER LABEL
When reconstituted in accordance with the package insert instructions, the concentration of aluminum will be no more than 42 m g/L.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
NASONEX (mometasone furoate) Nasal Spray
[July 17, 2002: Schering]
Labeling provides for the use of Nasonex (mometasone furoate) 50 mcg Nasal Spray in treatment of seasonal and perennial allergic rhinitis in children down to 2 years of age. Contact the company for a copy of the new labeling/package insert.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
OMNISCAN (gadodiamide) Injection
[July 8, 2002: Nycomed Amersham Imaging]
[Other labeling changes not found in 2002 PDR:
http://www.fda.gov/medwatch/SAFETY/2002/jun02.htm#omnisc ]
PRECAUTIONS
Geriatric Use
Of the total number of patients in clinical studies of OMNISCAN, 19.3 percent were 65 to 80, while 1.2 percent were over 80. No overall differences in safety or effectiveness were observed between these patients and younger patients, and other reported clinical experience has not identified differences in response between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out. In general, dose selection for an elderly patient should be cautious usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal or cardiac function, and of concomitant disease or other drug therapy.
This drug is known to be substantially excreted by the kidney, and the risk of toxic reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection, and it may be useful to monitor renal function.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
PERGONAL (menotropins) Injection
[July 30, 2002: Serono]
PRECAUTIONS
Geriatric Use
Clinical studies of Pergonal did not include sufficient numbers of male subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
PRAVACHOL (pravastatin sodium) Tablets
[July 12, 2002: Bristol-Myers Squibb]
ADVERSE REACTIONS
Adverse Clinical Events
Short-Term Controlled Trials
First paragraph revised:
All adverse clinical events (regardless of attribution) reported in more than 2% of pravastatin-treated patients in placebo-controlled trials of up to four months duration are identified in Table 6; also shown are the percentages of patients in whom these medical events were believed to be related or possibly related to the drug:
Table 6 - title added: "Adverse Events in > 2 Percent of Patients Treated with Pravastatin 10-40 mg in Short-Term Placebo-Controlled Trials."
Long-Term Controlled Morbidity and Mortality Trials
Adverse event data were pooled from seven double-blind, placebo-controlled trials (West of Scotland Coronary Prevention study [WOS]; Cholesterol and Recurrent Events study [CARE]; Long-term Intervention with Pravastatin in Ischemic Disease study [LIPID]; Pravastatin Limitation of Atherosclerosis in the Coronary Arteries study [PLAC I]; Pravastatin, Lipids and Atherosclerosis in the Carotids study [PLAC II]; Regression Growth Evaluation Statin Study [REGRESS]; and Kuopio Atherosclerosis Prevention Study [KAPS]) involving a total of 10,764 patients treated with pravastatin 40 mg and 10,719 patients treated with placebo. The safety and tolerability profile in the pravastatin group was comparable to that of the placebo group. Patients were exposed to pravastatin for a mean of 4.0 to 5.1 years in WOS, CARE, and LIPID and 1.9 to 2.9 years in PLAC I, PLAC II, KAPS, and REGRESS. In these long-term trials, the most common reasons for discontinuation were mild, non-specific gastrointestinal complaints. Collectively, these seven trials represent 47,613 patient-years of exposure to pravastatin. Events believed to be of probable, possible, or uncertain relationship to study drug, occurring in at least 1% of patients treated with pravastatin in these studies are identified in Table 7.
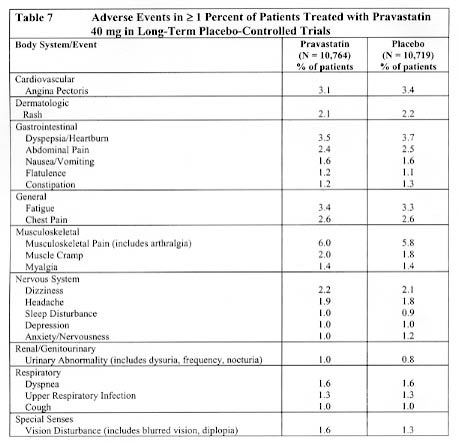
Events of probable, possible, or uncertain relationship to study drug that occurred in < 1.0% of pravastatin-treated patients in the long-term trials included the following; frequencies were similar in placebo-treated patients:
Dermatologic: pruritus, dermatitis, dryness skin, scalp hair abnormality (including alopecia), urticaria.
Endocrine/Metabolic: sexual dysfunction, libido change.
Gastrointestinal: decreased appetite.
General: fever, flushing.
Immunologic: allergy, edema head/neck.
Musculoskeletal: muscle weakness.
Nervous System: paresthesia, vertigo, insomnia, memory impairment, tremor, neuropathy (including peripheral neuropathy).
Special Senses: lens opacity, taste disturbance.
[Certain events below are deleted from the Postmarketing subsection because they now are included above in the Clinical Trials subsections.]
Postmarketing Experience
The following effects have been reported with drugs in this class; not all the effects listed below have necessarily been associated with pravastatin therapy:
In addition to the events reported above, as with other drugs in this class, the following events have been reported rarely during postmarketing experience with PRAVACHOL, regardless of causality assessment:
Skeletal Musculoskeletal: myopathy, rhabdomyolysis, arthralgia.
Neurological Nervous System: dysfunction of certain cranial nerves (including alteration of taste, impairment of extra-ocular movement, facial paresis), tremor, vertigo, memory loss, paresthesia, peripheral neuropathy, peripheral nerve palsy, anxiety, insomnia, depression.
Hypersensitivity Reactions: An apparent hypersensitivity syndrome has been reported rarely which has included one or more of the following features: anaphylaxis, angioedema, lupus erythematosus-like syndrome, polymyalgia rheumatica, dermatomyositis, vasculitis, purpura, thrombocytopenia, leukopenia, hemolytic anemia, positive ANA, ESR increase, eosinophilia, arthritis, arthralgia, urticaria, asthenia, photosensitivity, fever, chills, flushing, malaise, dyspnea, toxic epidermal necrolysis, erythema multiforme, including Stevens-Johnson syndrome.
Gastrointestinal: pancreatitis, hepatitis, including chronic active hepatitis, cholestatic jaundice, fatty change in liver, and, rarely, cirrhosis, fulminant hepatic necrosis, and hepatoma; anorexia, vomiting.
Skin Dermatologic: alopecia, pruritus. A variety of skin changes (e.g., nodules, discoloration, dryness of skin/mucous membranes, changes to hair/nails) have been reported.
Reproductive: gynecomastia, loss of libido, erectile dysfunction.
Eye: progression of cataracts (lens opacities), ophthalmoplegia.
Laboratory Abnormalities: elevated
transaminases,
alkaline phosphatase, and bilirubin; thyroid function abnormalities.
OVERDOSAGE
To date, there has been limited experience with overdosage of pravastatin. If an overdose occurs, it should be treated symptomatically with laboratory monitoring and supportive measures should be instituted as required. (See WARNINGS.)
STORAGE
Do not store above 86° F (30 ° C).
Store at 25° C (77° F); excursions permitted to 15° -30° C (59° - 86° F) [see USP Controlled Room Temperature]. Keep tightly closed (protect from moisture). Protect from light.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
PREVACID (lansoprazole) Delayed-Release Capsules
[July 31, 2002: TAP]
Labeling provides for the following changes in the Prevacid (lansoprazole) package insert: pediatric labeling for the 1 to 11 year age group by updating the Clinical Pharmacology, Clinical Studies, Indication and Usage, Precautions, Adverse Events, and Dosage and Administration sections. Contact the company for a copy of the new label/package insert.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
PRILOSEC (omeprazole) Delayed-Release Capsules
[July 15, 2002: AstraZeneca]
[Other labeling changes not in 2002 PDR: http://www.fda.gov/medwatch/SAFETY/2002/feb02.htm#prilos]
CLINICAL PHARMACOLOGY
Pharmacokinetics and Metabolism: Omeprazole
PRILOSEC Delayed-Release Capsule 40 mg was bioequivalent when administered with and without applesauce. However, PRILOSEC Delayed-Release Capsule 20 mg was not bioequivalent when administered with and without applesauce. When administered with applesauce, a mean 25% reduction in Cmax was observed without a significant change in AUC for PRILOSEC Delayed-Release Capsule 20 mg. The clinical relevance of this finding is unknown.
The pharmacokinetics of omeprazole have been investigated in pediatric patients of different ages.
Pharmacokinetic Parameters of Omeprazole Following Single and Repeated Oral Administration in Pediatric Populations Compared to Adults
|
Single or Repeated Oral Dosing/Parameter |
Children † <20 kg 2-5 years 10 mg |
Children† >20 kg 6-16 years 20 mg |
Adults‡ (mean 76 kg) 23-29 years (n=12) |
|
Single Dosing |
|||
|
Cmax* (ng/mL) |
288 (n=10) |
495 (n=49) |
668 |
|
AUC* (ng h/mL) |
511 (n=7) |
1140 (n=32) |
1220 |
|
Repeated Dosing |
|||
|
Cmax* (ng/mL) |
539 (n=4) |
851 (n=32) |
1458 |
|
AUC* (ng h/mL) |
1179 (n=2) |
2276 (n=23) |
3352 |
Note: * = plasma concentration adjusted to an oral dose of 1 mg/kg.
† Data from single and repeated dose studies
‡ Data from a single and repeated dose study
Doses of 10, 20 and 40 mg Omeprazole as Enteric-Coated Granules
Following comparable mg/kg doses of omeprazole, younger children (2-5 years) have lower AUCs than children 6 – 16 years or adults; AUCs of the latter two groups did not differ. (See DOSAGE AND ADMINISTRATION – Pediatric Patients.)
WARNINGS
Antimicrobials
Treatment with antibacterial agents alters the normal flora of the colon and may permit overgrowth of clostridia. Studies indicate that a toxin produced by Clostridium difficile is a primary cause of "antibiotic-associated colitis."
After the diagnosis of pseudomembranous colitis has been established, therapeutic measures should be initiated. Mild cases of pseudomembranous colitis usually respond to discontinuation of the drug alone. In moderate to severe cases, consideration should be given to management with fluids and electrolytes, protein supplementation, and treatment with an antibacterial drug clinically effective against Clostridium difficile colitis.
PRECAUTIONS
General
Information for Patients
Second paragraph -
For patients who have difficulty swallowing capsules, the contents of a PRILOSEC Delayed-Release Capsule can be added to applesauce. One tablespoon of applesauce should be added to an empty bowl and the capsule should be opened. All of the pellets inside the capsule should be carefully emptied on theapplesauce. The pellets should be mixed with the applesauce and then swallowed immediately with a glass of cool water to ensure complete swallowing of the pellets. The applesauce used should not be hot and should be soft enough to be swallowed without chewing. The pellets should not be chewed or crushed. The pellets/applesauce mixture should not be stored for future use.
Carcinogenesis, Mutagenesis, Impairment of Fertility
First paragraph, last sentence added:
A 26- week p53+/- transgenic mouse carcinogenicity study was not positive.
Pediatric Use
The safety and effectiveness of Prilosec have been established in the age group 2 years to 16 years for the treatment of acid-related gastrointestinal diseases, including the treatment of symptomatic GERD, treatment of erosive esophagitis, and the maintenance of healing of erosive esophagitis. The safety and effectiveness of Prilosec have not been established for pediatric patients less than 2 years of age. Use of Prilosec in the age group 2 years to 16 years is supported by evidence from adequate and well-controlled studies of Prilosec in adults with additional clinical, pharmacokinetic, and safety studies performed in pediatric patients (see CLINICAL PHARMACOLOGY, Pharmacokinetics and Metabolism: Omeprazole).
Treatment of Gastroesophageal Reflux Disease (GERD)
Symptomatic GERD
In an uncontrolled, open-label study of patients aged 2 years to 16 years with a history of symptoms suggestive of nonerosive GERD, 113 patients were assigned to receive a single daily dose of omeprazole (10 mg or 20 mg, based on body weight) either as intact capsule or as an open capsule in applesauce. Results showed success rates of 60% (10 mg omeprazole) and 59% (20 mg omeprazole) in reducing the number and intensity of either pain-related symptoms or vomiting/regurgitation episode was shown.
Erosive Esophagitis
In an uncontrolled, open-label dose-titration study, healing of erosive esophagitis in pediatric patients aged 1 to 16 years required doses that ranged from 0.7 to 3.5 mg/kg/day (80 mg/day). Doses were initiated at 0.7 mg/kg/day. Doses were increased in increments of 0.7 mg/kg/day (if intraesophageal pH showed a pH of < 4 for less than 6% of a 24-hour study). After titration, patients remained on treatment for 3 months. Forty-four percent of the patients were healed on a dose of 0.7 mg/kg body weight; most of the remaining patients were healed with 1.4 mg/kg after an additional 3 months’ treatment. Erosive esophagitis was healed in 51 of 57 (90%) children who completed the first course of treatment in the healing phase of the study. In addition, after 3 months of treatment, 33% of the children had no overall symptoms, 57% had mild reflux symptoms, and 40% had less frequent regurgitation/vomiting.
Maintenance of Healing of Erosive Esophagitis
In an uncontrolled, open-label study of maintenance of healing of erosive esophagitis in 46 pediatric patients, 54% of patients required half the healing dose. The remaining patients increased the healing dose (0.7 to a maximum of 2.8 mg/kg/day) either for the entire maintenance period, or returned to half the dose before completion. Of the 46 patients who entered the maintenance phase, 19 (41%) had no relapse. In addition, maintenance therapy in erosive esophagitis patients resulted in 63% of patients having no overall symptoms.
Safety
The safety of PRILOSEC Delayed-Release Capsules has been assessed in 310 pediatric patients aged 0 to 16 years and 62 physiologically normal volunteers aged 2 years to 16 years. Of the 310 pediatric patients with acid-related disease, a group of 46 who had documented healing of erosive esophagitis after 3 months of treatment continued on maintenance therapy for up to 749 days.
PRILOSEC Delayed-Release Capsules administered to pediatric patients was generally well tolerated with an adverse event profile resembling that in adults. Unique to the pediatric population, however, adverse events of the respiratory system were most frequently reported in both the 0 to 2 year and 2 to 16 year age groups (46.2% and 18.5%, respectively). Similarly, otitis media was frequently reported in the 0 to 2 year age group (22.6%), and accidental injuries were reported frequently in the 2 to 16 year age group (3.8%).
OVERDOSAGE
Rare Reports have been received of overdosage with omeprazole in humans. Doses ranged from 320 mg to 900 mg up to 2400 mg (16-45 120 times the usual recommended clinical dose). Manifestations were variable, but included confusion, drowsiness, blurred vision, tachycardia, nausea, vomiting, diaphoresis, flushing, headache, and dry mouth, and other adverse reactions similar to those seen in normal clinical experience. (See ADVERSE REACTIONS.) Symptoms were transient, and no serious clinical outcome has been reported when PRILOSEC was taken alone. No specific antidote for omeprazole overdosage is known. Omeprazole is extensively protein bound and is, therefore, not readily dialyzable. In the event of overdosage, treatment should be symptomatic and supportive.
As with the management of any overdose, the possibility of multiple drug ingestion should be considered. For current information on treatment of any drug overdose, a certified Regional Poison Control Center should be contacted. Telephone numbers are listed in the Physicians’ Desk Reference (PDR) or local telephone book.
Lethal doses of omeprazole after single oral administration are about 1500 mg/kg in mice and greater than 4000 mg/kg in rats, and about 100 mg/kg in mice and greater than 40 mg/kg in rats given single intravenous injections. Single oral doses of omeprazole at 1350, 1339, and 1200 mg/kg were lethal to mice, rats, and dogs, respectively. Animals given these doses showed sedation, ptosis, tremors, convulsions, and decreased activity, body temperature, and respiratory rate and increased depth of respiration.
DOSAGE AND ADMINISTRATION
Pediatric Patients
For the treatment of GERD or other acid-related disorders, the recommended dose for pediatric patients 2 years of age and older is as follows:
|
Patient Weight |
Omeprazole Dose |
|
<20kg |
10mg |
|
>20kg |
20mg |
On a per kg basis, the doses of omeprazole required to heal erosive esophagitis are greater than those for adults.
For pediatric patients unable to swallow an intact capsule see Alternative Administration Options subsection below.
Alternative Administration Options
For patients who have difficulty swallowing capsules, the contents of a PRILOSEC Delayed-Release Capsule can be added to applesauce. One tablespoon of applesauce should be added to an empty bowl and the capsule should be opened. All of the pellets inside the capsule should be carefully emptied on the applesauce. The pellets should be mixed with the applesauce and then swallowed immediately with a glass of cool water to ensure complete swallowing of the pellets. The applesauce used should not be hot and should be soft enough to be swallowed without chewing. The pellets should not be chewed or crushed. The pellet/applesauce mixture should not be stored for future use.
No dosage adjustment is necessary for patients with renal impairment or for the elderly.
PRILOSEC Delayed-Release Capsules should be taken before eating. In the clinical trials, antacids were used concomitantly with PRILOSEC.
Patients should be cautioned that the PRILOSEC Delayed-Release Capsule should not be opened, chewed or crushed, and should be swallowed whole.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
PROZAC (fluoxetine HCl) Capsules, Solution & Tablets
[July 7, 2002: Eli Lilly]
Labeling provides for the longer-term treatment of bulimia and the treatment of panic disorder, with or without agoraphobia. Contact the company for a copy of the new labeling/package insert.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
[July 14, 2002: Johnson & Johnson]
In double-blind, vehicle-controlled studies involving 339
patients who applied RENOVA 0.02% to their faces, adverse reactions associated
with the use of RENOVA were limited primarily to the skin. Almost all patients
reported one or more local reactions such as peeling, dry skin, burning, stinging,
erythema, and pruritus. In 32%
24%
of all study patients, skin irritation was reported that was either
severe (about 7%),
led to temporary discontinuation of RENOVA 0.02% (about
20%), or led to use of a mild topical corticosteroid.
About 7%
5%
of patients using RENOVA 0.02%, compared to less than 1% of the control patients,
had sufficiently severe local irritation to warrant short-term use of mild topical
corticosteroids to alleviate local irritation. About 4% of patients had to discontinue
use of RENOVA because of adverse reactions.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
SANDOSTATIN (octreotide acetate) Injection
[July 24, 2002: Novartis]
CLINICAL PHARMACOLOGY
Pharmacokinetics:
Second paragraph -
In healthy volunteers the distribution of octreotide from plasma was rapid (ta 1/2 = 0.2h), the volume of distribution (Vdss) was estimated to be 13.6L and the total body clearance was ranged from 7 L/hr to 10L/hr.
Last paragraph, last sentence of subsection:
The effect of hepatic diseases on the disposition of octreotide is unknown.
Patients with liver cirrhosis showed prolonged elimination of drug, with octreotide t 1/2 increasing to 3.7 hr and total body clearance decreasing to 5.9 L/hr, whereas patients with fatty liver disease showed t 1/2 increased to 3.4 hr and totally body clearance of 8.2 L/hr.
Pediatric Use
Single paragraph deleted from previous label and replaced with the following:
Experience with Sandostatin (octreotide acetate) in the pediatric population is limited.
Although formal controlled clinical trials have not been performed to evaluate safety and effectiveness in this age group, there are reports of 49 cases in the literature of neonates and infants with congenital hyperinsulinism [also called familial hyperinsulinism (HI), persistent hyperinsulinemic hypoglycemia of infancy (PHHI), or nesidioblastosis] who have received Sandostatin as an inhibitor of insulin release. The following efficacy and safety information is derived from these 49 patients.
Sandostatin has been used to stabilize plasma glucose levels prior to pancreatectomy and to treat recurrent post-operative hypoglycemia. Although most use of octreotide in this setting is short-term, a few reports in the literature have documented longer-term therapy in pediatric patients (2.2-5.5 years). Octreotide is an alternative medical treatment to diazoxide for control of hypoglycemia in this disorder. Of 31 pediatric patients who received Sandostatin as prescribed for congenital hyperinsulinism and for which long-term follow-up was available, octreotide obviated the need for surgery in 3 patients (10%) and was replaced by diazoxide in 4 patients (13%) due to uncontrolled hypoglycemia. Although the remainder of these patients required surgery, there have been a few reports in the literature of patients who have responded to octreotide after failing treatment with surgery and/or diazoxide. Doses of 3-40 mcg/kg/day have been used. At these doses, the majority of side effects were gastrointestinal: diarrhea, steatorrhea, vomiting, and abdominal distension, each reported in 22-35% (n = 11-17) of patients. However, they were generally short-lived – with resolution of vomiting and distention in 2-4 days, and diarrhea/steatorrhea, within 2-4 weeks. Steatorrhea was controlled in most patients with pancreatic enzyme supplements. Poor growth was reported in 37% of patients (n = 7) who received Sandostatin for 1-4.33 years. It was associated with low serum growth hormone and/or IGF-1 levels in 4/6 patients in whom these parameters were measured. Catch-up growth occurred in 3/3 patients who were followed after Sandostatin was discontinued. Poor weight gain was reported in 32% of patients (n = 6). Tachyphylaxis was reported in 35% (n = 17) of patients. Asymptomatic gallstones with sludge was reported in one infant after one year of therapy and was treated with ursodeoxycholic acid. There has been a single report of an infant with nesidioblastosis who experienced a seizure thought to be independent of Sandostatin therapy. A single death has been reported in a 16-month-old male with enterocutaneous fistula who developed sudden abdominal pain and increased nasogastric drainage and expired 8 hours after receiving a single 100 mcg subcutaneous dose of Sandostatin.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
SPORANOX (itraconazole) Capsules, Oral Solution & Injection
[July 17, 2002: Johnson and Johnson]
[Other labeling changes not in 2002 PDR: http://www.fda.gov/medwatch/SAFETY/2002/feb02.htm#sporan
http://www.fda.gov/medwatch/SAFETY/2002/apr02.htm#sporan]
WARNINGS
Second paragraph is now bolded and revised:
Hepatic Effects: SPORANOX has been associated with rare cases of serious
hepatotoxicity, including liver failure and death. Some of these cases had neither pre-existing liver disease nor a serious underlying medical condition and some of these cases developed within the first week of treatment. If clinical signs or symptoms develop that are consistent with liver disease, the risks and benefits of continued treatment should be discontinued and liver function testing performed. Continued SPORANOX use should be reassessed. or reinstitution of treatment with SPORANOX is strongly discouraged unless there is a serious or life threatening situation where the expected benefit exceeds the risk.
(See PRECAUTIONS: Information for Patients and ADVERSE [Oral Solution label] EVENTS REACTIONS REACTIONS.)
PRECAUTIONS
General:
Hepatic enzyme test values should be monitored in patients with pre-existing hepatic function abnormalities or those who have experienced liver toxicity with other medications. Hepatic enzyme test values should be monitored periodically in all patients receiving continuous treatment for more than 1 month, or at any time a patient develops signs or symptoms suggestive of liver dysfunction.
Rare cases of serious hepatotoxicity have been observed with Sporanox treatment, including some cases within the first week. In patients with elevated or abnormal liver enzymes or active liver disease, or who have experienced liver toxicity with other drugs, treatment with Sporanox is strongly discouraged unless there is a serious or life threatening situation where the expected benefit exceeds the risk. Liver function monitoring should be done in patients with pre-existing hepatic function abnormalities or those who have experienced liver toxicity with other medications and should be considered in all patients receiving Sporanox. Treatment should be stopped immediately and liver function testing should be conducted in patients who develop signs and symptoms suggestive of liver dysfunction.
If neuropathy occurs that may be attributable to Sporanox capsules, the treatment should be discontinued.
[Capsule and Oral Solution labels]
Information for Patients
Fourth bullet revised -
· Instruct patients to report stop Sporanox treatment immediately and contact their healthcare provider if any signs and symptoms that may suggest suggestive of liver dysfunction develop. Such signs and symptoms may include unusual fatigue, anorexia, nausea and/or vomiting, jaundice, dark urine, or pale stools.
[Capsule and Oral Solution labels]
Pediatric Use
A small number of patients ages 3 to 16 years have been treated with 100 mg/day of itraconazole capsules for systemic fungal infections, and no serious unexpected adverse effects events have been reported.
ADVERSE REACTIONS
First paragraph, third sentence revised:
If clinical signs or symptoms develop that are consistent with liver disease, the treatment should be discontinued and liver function testing performed. The risks and benefits of continued SPORANOXuse should be reassessed. (See WARNINGS: Hepatic Effects and PRECAUTIONS: General and Information for Patients.)
The title of the first Adverse Events table in the section was changed from "Treatment of Systemic Fungal Infections" to "Clinical Trials of Systemic Fungal Infections".
Post-marketing Experience
In worldwide post-marketing experience with SPORANOXCapsules in treatment of onychomycosis and/or systemic fungal infections, peripheral edema, allergic reactions, including rash, pruritus, urticaria, angioedema, and, in rare instances, anaphylaxis and Stevens-Johnson syndrome, have been reported. Post-marketing experiences have also included reports of elevated liver enzymes and rarely liver failure. Rare cases of congestive heart failure and pulmonary edema have been reported. Rare cases of alopecia, hypertriglyceridemia, menstrual disorders, and neutropenia, and isolated cases of neuropathy have also been reported, although the causal association with SPORANOX is uncertain. (See CLINICAL PHARMACOLOGY: Special Populations, CONTRAINDICATIONS, WARNINGS, and PRECAUTIONS: Drug Interactions for more information).
Worldwide post-marketing experiences with the use of SPORANOX include adverse events of gastrointestinal origin, such as dyspepsia, nausea, vomiting, diarrhea, abdominal pain and constipation. Other reported adverse events include peripheral edema, congestive heart failure and pulmonary edema, headache, dizziness, peripheral neuropathy, menstrual disorders, reversible increases in hepatic enzymes, hepatitis, liver failure, hypokalemia, hypertriglyceridemia, alopecia, allergic reactions (such as pruritus, rash, urticaria, angioedema, anaphylaxis), Stevens-Johnson syndrome, and neutropenia. (See CLINICAL PHARMACOLOGY: Special Populations, CONTRAINDICATIONS, WARNINGS, and PRECAUTIONS: Drug Interactions for more information).
[Capsules label]
Patient Package Insert (PPI)
• lovastatin (such as Mevacor, Advicor)
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
TAXOTERE (docetaxel) Injection
[July 9 & 30, 2002: Aventis]
ADVERSE REACTIONS
Post-marketing Experiences
Ongoing Evaluation: The following serious adverse events of uncertain relationship to TAXOTERE have been reported:
The following adverse events have been identified from clinical trials and/or post-marketing surveillance. Because they are reported from a population of unknown size, precise estimates of frequency cannot be made.
Body as a whole: abdominal pain, diffuse pain, chest pain, radiation recall phenomenon
Cardiovascular: atrial fibrillation, deep vein thrombosis, ECG abnormalities, thrombophlebitis, pulmonary embolism, syncope, tachycardia, myocardial infarction
Cutaneous: rare cases of bullous eruption such as erythema multiforme or Stevens-Johnson syndrome. Multiple factors may have contributed to the development of these effects.
Gastrointestinal Digestive: abdominal pain, anorexia, constipation, duodenal ulcer, esophagitis, gastrointestinal hemorrhage, gastrointestinal perforation, ischemic colitis, colitis, intestinal obstruction, ileus, neutropenic enterocolitis and dehydration in relation to digestive disorders as a consequence to gastrointestinal events have been reported.
Hematologic: bleeding episodes
Hepatic: rare cases of hepatitis have been reported.
Neurologic Nervous: confusion, seizures rare cases of seizures or transient loss of consciousness have been observed, sometimes appearing during the infusion of the drug.
Ophthalmologic: conjunctivitis, lacrimation or lacrimation with or without conjunctivitis. Excessive tearing which may be attributable to lacrimal duct obstruction has been reported.
Respiratory: dyspnea, acute pulmonary edema, acute respiratory distress syndrome, interstitial pneumonia
Urogenital: renal insufficiency
PREPARATION AND ADMINISTRATION PRECAUTIONS
A. Preparation of the Initial Diluted Solution
2. Aseptically withdraw the entire contents of the appropriate diluent vial into a syringe by partially inverting the vial, and transfer it to the appropriate vial of TAXOTERE for Injection Concentrate. If the procedure is followed as described, an initial diluted solution of 10mg docetaxel/mL will result.
3. Gently
rotate Mix
the initial diluted solution by
repeated inversions for
approximately at
least 15
45
seconds to assure full mixture of the concentrate
and diluent. Do not shake.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
TESLASCAN (mangafodipir trisodium) Injection
[July 8, 2002: Nycomed Amersham Imaging]
PRECAUTIONS
Geriatric Use
Of the total number of patients in eight Phase 1, 2 and 3 clinical studies of TESLASCAN, 218 of 678 (32.2%) were 65 to 80 years old, while 8 of 678 (1.2%) were over 80 years old. No overall differences in safety or effectiveness were observed between these patients and younger patients, and other reported clinical experience has not identified differences in response between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out. In general, dose selection for an elderly patient should be cautious usuallystarting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal or cardiac function, and of concomitant disease or other drug therapy.
This drug is known to be substantially excreted by the kidney, and the risk of toxic reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection, and it may be useful to monitor renal function.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
ZITHROMAX (azitrhomycin dihydrate) Capsules,
Oral Suspension,
Tablets, Single Dose Packet & Injection
[July 22, 2002: Pfizer]
[Other labeling changes not in 2002 PDR: http://www.fda.gov/medwatch/safety/2001/dec01.htm#zithro ,
http://www.fda.gov/medwatch/SAFETY/2002/may02.htm#zithro ]
PRECAUTIONS
General:
Second paragraph -
The following adverse events have not been reported in clinical trails with azithromycin, an azalide; however, they have been reported with macrolides products: ventricular arrhythmias, including ventricular tachycardia and torsade de pointes, in individuals with prolong QT intervals.
The following adverse events have been reported with macrolide products: ventricular arrhythmias, including ventricular tachycardia and torsade de pointes, in individuals with prolonged QT intervals.
ADVERSE REACTIONS
In clinical trials, most of the reported side effects were mild to moderate in severity and were reversible upon discontinuation of the drug. Potentially serious side effects of angioedema and cholestatic jaundice were reported rarely. Approximately 0.7% of the patients (adults and children) from the 5-day multiple-dose clinical trials discontinued ZITHROMAX (azithromycin) therapy because of treatment-related side effects. In clinical trials in children given 30 mg/kg, either as a single dose or over 3 days, discontinuation from the trials due to treatment-related side effects was approximately 1%. (See DOSAGE AND ADMINISTRATION.) Most of the side effects leading to discontinuation were related to the gastrointestinal tract, e.g., nausea, vomiting, diarrhea, or abdominal pain. (See CLINICAL STUDIES IN PEDIATRIC PATIENTS.) Potentially serious side effects of angioedema and cholestatic jaundice were reported rarely.
Clinical:
Children:
Single and Multiple-dose regimens: The types of side effects in children were comparable to those seen in adults, with different incidence rates for the two dosage regimens recommended in children.
Acute Otitis Media: For the recommended total dosage regimen of 30 mg/kg, the most frequent side effects ³ 1%) attributed to treatment were diarrhea, abdominal pain, vomiting, nausea and rash. (See DOSAGE AND ADMINISTRATION, CLINICAL STUDIES IN PEDIATRIC PATIENTS.) The incidence, based on dosing regimen, is described in the table below:
|
Dosage Regimen |
Diarrhea,% |
Abdominal Pain,% |
Vomiting,% |
Nausea, % |
Rash, % |
|
1-day |
4.3% |
1.4% |
4.9% |
1.0% |
1.0% |
|
3-day |
2.6% |
1.7% |
2.3% |
0.4% |
0.6% |
|
5-day |
1.8% |
1.2% |
1.1% |
0.5% |
0.4% |
Community-Acquired Pneumonia: For the recommended dosage regimen of 10 mg/kg on Day 1 followed by 5 mg/kg on Days 2-5, the most frequent side effects attributed to treatment were diarrhea/loose stools, abdominal pain, vomiting, nausea, and rash.
The incidence is described in the table below:
|
Dosage Regimen |
Diarrhea/ Loose stools, % |
Abdominal Pain,% |
Vomiting,% |
Nausea, % |
Rash, % |
|
5-day |
5.8% |
1.9% |
1.9% |
1.9% |
1.6% |
Pharyngitis/tonsillitis: For the recommended dosage regimen of 12 mg/kg on Days 1-5, the most frequent side effects attributed to treatment were diarrhea/loose stools, vomiting, abdominal pain, nausea, and headache.
The incidence is described in the table below:
|
Dose Regimen |
Diarrhea,% |
Vomiting,% |
Abdominal Pain, % |
Nausea,% |
Rash,% |
Headache,% |
|
5-day |
5.4% |
5.6% |
3.4% |
1.8% |
0.7% |
1.1% |
With either any of the treatment regimens, no other treatment-related side effects occurred in children treated with ZITHROMAX with a frequency greater than 1%. Side effects that occurred with a frequency of 1% or less included the following:
Cardiovascular: Chest pain.
Gastrointestinal: Dyspepsia, constipation, anorexia, enteritis, flatulence, gastritis, jaundice, loose stools and oral moniliasis.
Hematologic and Lymphatic: anemia and leukopenia.
Nervous System: Headache (otitis media dosage), hyperkinesia, dizziness, agitation, nervousness and insomnia.
General: Fever, face edema, fatigue, fungal infection, malaise and pain.
Allergic: Rash and allergic reaction.
Respiratory: Cough increased, pharyngitis, pleural effusion and rhinitis.
Skin and Appendages: Eczema, fungal dermatitis, pruritus, sweating, urticaria and vesiculobullous rash.
Special Senses: Conjunctivitis.
Post-Marketing Experience:
Adverse events reported with azithromycin during the post-marketing period in adult and/or pediatric patients for which a causal relationship may not be established include:
Allergic: Arthralgia, edema, urticaria and angioedema.
Cardiovascular: Arrhythmias including ventricular tachycardia and hypotension.
Gastrointestinal: Anorexia, constipation, dyspepsia, flatulence, vomiting/diarrhea rarely resulting in dehydration, pseudomembranous colitis, pancreatitis, oral candidiasis and rare reports of tongue discoloration.
General: Asthenia, paresthesia, fatigue, malaise and anaphylaxis (rarely fatal).
Genitourinary: Interstitial nephritis and acute renal failure, oral candidiasis and vaginitis.
Hematopoietic: Thrombocytopenia.
Liver/Biliary: Abnormal liver function including hepatitis and cholestatic jaundice, as well as rare cases of hepatic necrosis and hepatic failure, some of which have rarely resulted in death.
Nervous System: Convulsions, dizziness/vertigo, headache, somnolence, hyperactivity, nervousness agitation, and syncope.
Psychiatric: Aggressive reaction and anxiety.
Skin/Appendages: Pruritus, rarely serious skin reactions including erythema multiforme, Stevens-Johnson syndrome and toxic epidermal necrolysis.
Special Senses: Hearing disturbances including hearing loss, deafness and/or tinnitus and rare reports of taste perversion.
Laboratory Abnormalities:
Children:
Previous text replaced by the following:
One, Three and Five Day Regimens
In multiple-dose clinical trials involving approximately 4700 pediatric patients, no patients discontinued therapy because of treatment-related laboratory abnormalities.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
HTML by JLW