

|
|
(Posted: 2/20/2002)
How to Find a Safety-Related Labeling ChangeUse the drop-down menu to select a product by brand name. |
MedWatch
Home | What's New | About
Medwatch | How to Report | Submit
Report | Safety Info
Continuing Education | Download
PDF | Comments | Privacy
Statement
ACTOS (pioglitazone) Tablets
[January 7, 2002: Takeda Pharmaceuticals North America]
[Other labeling changes not appearing in 2001 PDR: http://www.fda.gov/medwatch/SAFETY/2001/jul01.htm#actos ]
CLINICAL PHARMACOLOGY
Pharmacodynamics and Clinical Effects moved to PRECAUTIONS, under a new section heading Weight Gain. The information was assembled in tabular form and included medians, 25% and 75% percentiles.
In all clinical trials, a reduction in HbA1c was accompanied by increased body weight in patients treated with ACTOS in a dose-related manner. The change in average weight in U.S. placebo-controlled monotherapy trials ranged from 0.5 kg to 2.8 kg for patients treated with ACTOS and -1.3 kg to -1.9 kg for placebo-treated patients. In combination with sulfonylurea, the change in average weight was 1.9 kg and 2.9 kg for 15 mg and 30 mg of ACTOS, respectively, and -0.8 kg for placebo. In combination with insulin, the change in average weight was 2.3 kg and 3.7 kg for 15 mg and 30 mg of ACTOS, respectively, and 0kg for placebo. In combination with metformin, the change in average weight was 1.0 kg for 30 mg of ACTOS and -1.4 kg for placebo.
[See PRECAUTIONS below]
WARNINGS
Cardiac Failure and Other Cardiac Effects
ACTOS, like other thiazolidinediones, can cause fluid retention when used alone or in combination with other antidiabetic agents, including insulin. Fluid retention may lead to or exacerbate heart failure. Patients should be observed for signs and symptoms of heart failure (see Information for Patients).
ACTOS should be discontinued if any deterioration in cardiac status occurs. Patients with New York Heart Association (NYHA) Class III and IV cardiac status were not studied during clinical trials; therefore, ACTOS is not recommended in these patients (see PRECAUTIONS, Cardiovascular).
In one 16-week U.S. double-blind, placebo-controlled clinical trial involving 566 patients with type 2 diabetes, ACTOS at doses of 15 mg and 30 mg in combination with insulin were compared to insulin therapy alone. This trial included patients with long-standing diabetes and a high prevalence of pre-existing medical conditions as follows: arterial hypertension (57.2%), peripheral neuropathy (22.6%), coronary heart disease (19.6%), retinopathy (13.1%), myocardial infarction (8.8%), vascular disease (6.4%), angina pectoris (4.4%), stroke and/or transient ischemic attack (4.1%), and congestive heart failure (2.3%).
In this study two of the 191 patients receiving 15 mg ACTOS plus insulin (1.1%) and two of the 188 patients receiving 30 mg ACTOS plus insulin (1.1%) developed congestive heart failure compared with none of the 187 patients on insulin therapy alone. All four of these patients had previous histories of cardiovascular conditions including coronary artery disease, previous CABG procedures, and myocardial infarction.
Analysis of data from this study did not identify specific factors that predict increased risk of congestive heart failure on combination therapy with insulin.
PRECAUTIONS
Cardiac section deleted and the following new Cardiovascular section included:
Cardiovascular : In U.S. placebo-controlled clinical trials that excluded patients with New York Heart Association (NYHA) Class III and IV cardiac status, the incidence of serious cardiac adverse events related to volume expansion was not increased in patients treated with ACTOS as monotherapy or in combination with sulfonylureas or metformin vs. placebo-treated patients. In insulin combination studies, a small number of patients with a history of previously existing cardiac disease developed congestive heart failure when treated with ACTOS in combination with insulin (see WARNINGS). Patients with NYHA Class III and IV cardiac status were not studied in ACTOS clinical trials. ACTOS is not indicated in patients with NYHA Class III or IV cardiac status.
In postmarketing experience with ACTOS, cases of congestive heart failure have been reported in patients both with and without previously known heart disease.
PRECAUTIONS
Edema
In double blind clinical trials of patients with type 2 diabetes, mild to moderate edema was reported in patients treated with ACTOS.
In all U.S. clinical trials, edema was reported more frequently in patients treated with ACTOS than in placebo-treated patients (see ADVERSE REACTIONS). In postmarketing experience, reports of initiation or worsening of edema have been received.
Weight Gain - Dose related weight gain was seen with ACTOS alone and in combination with other hypoglycemic agents (Table 6). The mechanism of weight gain is unclear but probably involves a combination of fluid retention and fat accumulation.
|
Table 6 Weight Changes (kg) from Baseline during Double-Blind Clinical Trials with ACTOS |
||||||
|
Control Group (Placebo) |
ACTOS 15 mg |
ACTOS 30 mg |
ACTOS 45 mg |
|||
|
Median (25th/75th percentile) |
Median (25th/75th percentile) |
Median (25th/75th percentile) |
Median (25th/75th percentile) |
|||
|
Monotherapy |
-1.4 n=256 |
0.9 n=79 |
1.0 n=188 |
2.6 n=79 |
||
|
Combination Therapy |
Sulfonylurea |
-0.5 n=187 |
2.0 n=183 |
2.7 n=186 |
N/A |
|
|
Metformin |
-1.4 n=160 |
N/A |
1.4 n=167 |
N/A |
||
|
Insulin |
0.2 n=182 |
2.3 n=190 |
3.6 n=188 |
N/A |
||
Information for Patients
Patients who experience an unusually rapid increase in weight or edema or who develop shortness of breath or other symptoms of heart failure while on ACTOS should immediately report these symptoms to their physician.
ADVERSE REACTIONS
The types of clinical adverse events reported when ACTOS was used in combination with sulfonylureas (N=373), metformin (N=168), or insulin (N=379) were generally similar to those reported during ACTOS monotherapy with the exception of an increase in the occurrence of edema in the insulin combination study (pioglitazone 15%and placebo 7%)
For most clinical adverse events the incidence was similar for groups treated with ACTOS monotherapy and those treated in combination with sulfonylureas, metformin, and insulin. There was an increase in the occurrence of edema in the patients treated with ACTOS and insulin compared to insulin alone.
In the ACTOS plus insulin trial (n=379), 10 patients treated with ACTOS plus insulin developed dyspnea and also, at some point during the therapy developed either weight change or edema. Seven of these 10 patients received diuretics to treat these symptoms. This was not reported in the insulin plus placebo group.
In all U.S. clinical trials, edema was reported more frequently in patients treated with ACTOS than in placebo-treated patients.
In combination therapy studies, edema was reported for 7.2% of patients treated with ACTOS and sulfonylureas compared to 2.1% of patients on sulfonylureas alone. In combination therapy studies with metformin, edema was reported in 6.0% of patients on combination therapy compared to 2.5% of patients on metformin alone. In combination therapy studies with insulin, edema was reported in 15.3% of patients on combination therapy compared to 7.0% of patients on insulin alone (see PRECAUTIONS, Edema).All Most of these events were considered mild or moderate in intensity.
In one 16-week clinical trial of insulin plus ACTOS combination therapy, more patients developed congestive heart failure on combination therapy (1.1%) compared to none on insulin alone (see WARNINGS, Cardiac Failure and Other Cardiac Effects).
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
ALKERAN (mephalan HCl) Tablets & Injection
[January 7, 2002: GlaxoSmithKline]
ADVERSE REACTIONS:
Gastrointestinal:
Gastrointestinal disturbances such as nausea and vomiting, diarrhea, and oral ulceration occur infrequently. Hepatic disorders ranging from abnormal liver function tests to clinical manifestations such as hepatitis and jaundice have been reported.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
CAFCIT (caffeine citrate) Injection & Oral Solution
[January 18, 2002: O.P.R. Development, L.P. c/o Roxane Laboratories]
CLINICAL PHARMACOLOGY
Pharmacokinetics
Special Populations: Studies examining the pharmacokinetics of caffeine in neonates with hepatic or renal insufficiency have not been conducted. CAFCIT (caffeine citrate) should be administered with caution in preterm neonates with impaired renal or hepatic function.
Serum concentrations of caffeine should be monitored and dose administration of CAFCIT should be adjusted to avoid toxicity in this population.
PRECAUTIONS
Renal and hepatic Systems
CAFCITshould be administered with caution in infants with impaired renal or hepatic function. Serum concentrations of caffeine should be monitored and dose administration of CAFCIT should be adjusted to avoid toxicity in this population. (See CLINICAL PHARMACOLOGY, Elimination, Special Populations).
OVERDOSAGE
Following overdosage, serum caffeine levels have ranged from approximately 24 mg/L (a post-marketing spontaneous case report in which an infant exhibited irritability, poor feeding and insomnia) to 350 mg/L. Serious toxicity has been associated with serum levels greater than 50 mg/L (see PRECAUTIONS-Laboratory tests and DOSAGE ADMINISTRATION). Signs and symptoms reported in the literature after caffeine overdosage in preterm infants include fever, tachypnea, jitteriness, insomnia, fine tremor of the extremities, hypertonia, opisthotonos, tonic-clonic movements, nonpurposeful jaw and lip movements, vomiting, hyperglycemia, elevated blood urea nitrogen, and elevated total leukocyte concentration. Seizures have also been reported in cases of overdose. One case of caffeine overdose complicated by development of intraventricular hemorrhage and long-term neurologic sequelae has been reported. Another case
of caffeine citrate overdose (from New Zealand; not CAFCIT) of an estimated 600 mg caffeine citrate (approximately 322 mg/kg) administered over 40 minutes was complicated by tachycardia, ST depression, respiratory distress, heart failure, gastric distention, acidosis, and a severe extravasation burn with tissue necrosis at the peripheral intravenous injection site. No deaths associated with caffeine overdose have been reported in preterm infants.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
[January 23, 2002: Pfizer]
ADVERSE REACTIONS
Added to the post-marketing experience section:
Skin Disorders: uticaria
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
CLAFORAN (cefotaxime sodium) Injection and Sterile Solution
[January 18, 2002: Aventis]
PRECAUTIONS
Carcinogenesis, Mutagenesis, and Impairment of Fertility: Lifetime studies in animals to evaluate carcinogenic potential have not been conducted. Claforan was not mutagenic in the mouse micronucleus test or in the Ame's test. Claforan did not impair fertility to rats when administered subcutaneously at doses up to 250 mg/kg/day (0.2 times the maximum recommended human dose based on mg/m2 ) or in mice when administered intravenously at doses up to 2000 mg/kg/day (0.7 times the recommended human dose based on mg/m2).
Pregnancy: Teratogenic Effects: Pregnancy Category B: Reproduction studies have been performed in pregnant mice given Claforan intravenously at doses up to 1200 mg/kg/day (0.4 times the recommended human dose based on mg/m2) or in pregnant rats when administered intravenously at doses up to 1200 mg/kg/day (0.8 times the recommended human dose based on mg/m2). No evidence of embryotoxicity or teratogenicity was seen in these studies.
ADVERSE REACTIONS Added: a list of cutaneous adverse reactions (erythema multiforme, Stevens-Johnson syndrome, and toxic epidermal necrolysis).
OVERDOSAGE
The acute toxicity of Claforan was evaluated in neonatal and adult mice and rats. Significant mortality was seen at parenteral doses in excess of 6000 mg/kg/day in all groups. Common toxic signs in animals that died were a decrease in spontaneous activity, tonic and clonic convulsions, dyspnea, hypothermia, and cyanosis.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
[January 14, 2002: Novartis Pharmaceuticals]
[Other labeling changes not appearing in 2001 PDR: http://www.fda.gov/medwatch/SAFETY/2001/aug01.htm#clozar
]
Dear Healthcare Professional Letter (Feb 2002): http://www.fda.gov/medwatch/SAFETY/2002/Clozaril_deardoc.pdf
Revised Package Insert (Feb 2002):
http://www.fda.gov/medwatch/SAFETY/2002/Clozaril_PI.pdf
The previously existing BOXED WARNING has been relocated to the beginning of the package insert and revised to advise health care providers of the association of myocarditis with clozapine therapy. A subsection has been added to the WARNINGS section entitled "Myocarditis" to provide data and clozapine treatment guidelines related to this issue.
|
3. MYOCARDITIS ANALYSES OF POSTMARKETING SAFETY DATABASES SUGGEST THAT CLOZAPINE IS ASSOCIATED WITH AN INCREASED RISK OF FATAL MYOCARDITIS, ESPECIALLY DURING, BUT NOT LIMITED TO, THE FIRST MONTH OF THERAPY. IN PATIENTS IN WHOM MYOCARDITIS IS SUSPECTED, CLOZAPINE TREATMENT SHOULD BE PROMPTLY DlSCONTINUED. (SEE WARNINGS) |
WARNINGS
Myocarditis
Post-marketing surveillance data from four countries that employ hematological monitoring of clozapine-treated patients revealed: 30 reports of myocarditis with 17 fatalities in 205,493 U.S. patients (August 2001); 7 reports of myocarditis with 1 fatality in 15,600 Canadian patients (April 2001); 30 reports of myocarditis with 8 fatalities in 24,108 U.K. patients (August 2001); 15 reports of myocarditis with 5 fatalities in 8,000 Australian patients (March 1999). These reports represent an incidence of 5.0, 16.3, 43.2, and 96.6 cases/100,000 patient years, respectively. The number of fatalities represent an incidence of 2.8, 2.3, 11.5, and 32.2 cases/100,000 patient years, respectively.
The overall incidence rate of myocarditis in patients with schizophrenia treated with antipsychotic agents is unknown. However, for the established market economies (WHO), the incidence of myocarditis is 0.3 cases/100,000 patient years and the fatality rate is 0.2 cases/100,000 patient years. Therefore, the rate of myocarditis in clozapine treated patients appears to be 17-322 times greater than the general population and is associated with an increased risk of fatal myocarditis that is 14-161 times greater than the general population.
The total reports of myocarditis for these four countries was 82 of which 51 (62%) occurred within the first month of clozapine treatment, 25 (31%) occurred after the first month of therapy and 6 (7%) were unknown. The median duration of treatment was 3 weeks. Of 5 patients rechallenged with clozapine, 3 had a recurrence of myocarditis. Of the 82 reports, 31 (38%) were fatal and 25 patients who died had evidence of myocarditis at autopsy. These data also suggest that the incidence of fatal myocarditis may be highest during the first month of therapy.
Therefore, the possibility of myocarditis should be considered in patients receiving Clozaril (clozapine) who present with unexplained fatigue, dyspnea, tachypnea, fever, chest pain, palpitations, other signs or symptoms of heart failure, or electrocardiographic findings such as ST- T wave abnormalities or arrhythmias. It is not known whether eosinophilia is a reliable predictor of myocarditis. Tachycardia, which has been associated with Clozaril (Clozapine) treatment, has also been noted as a presenting sign in patients with myocarditis. Therefore, tachycardia during the first month of therapy warrants close monitoring for other signs of myocarditis.
Prompt discontinuation of Clozaril (clozapine) treatment is warranted upon suspicion of myocarditis. Patients with clozapine-related myocarditis should not be rechallenged with Clozaril (clozapine).
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
[January 25, 2002: Merck]
[Other labeling changes not appearing in 2001 PDR: http://www.fda.gov/medwatch/SAFETY/2001/mar01.htm#crixiv ]
ADVERSE REACTIONS:
Clinical Trials in Adults
First paragraph changed from:
Nephrolithiasis/urolithiasis, including flank pain with or without hematuria (including microscopic hematuria), has been reported in approximately 9.3% (193/2071) of patients receiving CRIXIVAN in clinical trials at the recommended dose, compared to 1.8% in the control arms. Of the patients treated with CRIXIVAN who developed nephrolithiasis/urolithiasis, 3.1% (6/193) were reported to develop hydronephrosis and 3.1% (6/193) underwent stent placement. Following the acute episode, 3.6% (7/193) of patients discontinued therapy. (See WARNINGS and DOSAGE AND ADMINISTRATION, Nephrolithiasis/Urolithiasis.)
To the following:
Nephrolithiasis/urolithiasis, including flank pain with or without hematuria (including microscopic hematuria), has been reported in approximately 12.4% (301/2429; range across individual trials: 4.7% to 34.4%) of patients receiving CRIXIVAN at the recommended dose in clinical trials with a median follow-up of 47 weeks (range: 1 day to 242 weeks; 2238 patient-years follow-up). The cumulative frequency of nephrolithiasis events increases with duration of exposure to CRIXIVAN; however, the risk over time remains relatively constant. Of the patients treated with CRIXIVAN who developed nephrolithiasis/urolithiasis in clinical trials during the double-blind phase, 2.8% (7/246) were reported to develop hydronephrosis and 4.5% (11/246) underwent stent placement. Following the acute episode, 4.9% (12/246) of patients discontinued therapy. (See WARNINGS and DOSAGE AND ADMINISTRATION, Nephrolithiasis/Urolithiasis.)
ADVERSE REACTIONS:
Urogenital System - pyelonephritis with or without bacteremia was added.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
DAYPRO (oxaprozin) Caplets
[January 25, 2002: G.D. Searle]
CLINICAL PHARMACOLOGY
Pharmacokinetics
Excretion: Biliary excretion of unchanged oxaprozin is a minor pathway, and enterohepatic recycling of oxaprozin is insignificant.
CONTRAINDICATIONS
Daypro is contraindicated in patients with known hypersensitivity to oxaprozin. Daypro should not be given to patients who have experienced asthma, urticaria, or allergic-type reactions after taking aspirin or other NSAIDs.
PRECAUTIONS
Drug interactions
ACE-inhibitors: Reports suggest that NSAMs may diminish the antihypertensive effect of ACE-inhibitors. Oxaprozin has been shown to alter the pharmacokinetics of enalapril (significant decrease in dose-adjusted AUC 0-24 and Cmax and its active metabolite enalaprilat (significant increase in dose-adjusted AUC 0-24 . This interaction should be given consideration in patients taking NSAIDs concomitantly with ACE-inhibitors.
Pediatric use:
Second paragraph -
The pharmacokinetic profile and tolerability of oxaprozin were assessed in IRA patients 6-16 years of age relative to adult rheumatoid arthritis patients in a 1.4 day multiple dose pharmacokinetic study. Apparent clearance of unbound oxaprozin in IRA
patients was reduced compared to adult rheumatoid arthritis patients, but this reduction could be accounted for by differences in body weight (see Pharmacokinetics: Pediatric patients). No pharmacokinetic data are available for pediatric patients under 6 years. Adverse events were reported by approximately 45% of IRA patients and were somewhat more frequent when compared to the versus an approximate 30% incidence of adverse events in the adult rheumatoid arthritis patient cohort. Most 4f the adverse events were related to the gastrointestinal tract and were mild to moderate.
Third paragraph, first sentence -
In a 3 month open label study, 10 - 20 mg/kg/day of oxaprozin were administered to 59 IRA patients aged 3-16 years.
Geriatric use:
Second paragraph, first sentence -
Of the total number of subjects evaluated in four placebo controlled and active clinical studies of oxaprozin, 34 39% were 65 and over, and 11% were 75 and over.
New fourth paragraph -
Daypro is substantially excreted by the kidney, and the risk of toxic reactions to Daypro may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection, and it may be useful to monitor renal function (see PRECAUTIONS Renal Effects).
ADVERSE REACTIONS
INCIDENCE LESS THAN 1 %
The following adverse reactions were reported in clinical trials, from worldwide marketing experience (in italics) or in patients taking other NSAIDs (double asterisks**).
Cardiovascular system: edema, blood pressure changes, congestive heart failure**, hypertension**, palpitations, tachycardia**, syncope**.
Hematologic system: agranulocytosis, anemia, ecchymosis, eosinophilia **, melena**, pancyropenia, purpura**, thrombocytopenia, leukopenia.
Metabolic system: weight changes.
Nervous system: anxiety**, asthenia**, confusion**, depression**, dream abnormalities**, drowsiness**, insomnia**, malaise, nervousness**, paresthesia**, somnolence**, tremors**, vertigo**, weakness.
Respiratory system: asthma**, dyspnea**, pulmonary infections, pneumonia**, sinusitis, symptoms of upper respiratory tract infection, respiratory depression**.
Skin: alopecia, angioedema**, pruritus, urticaria, photosensitivity, pseudoporphyria, exfoliative dermatitis, erythema multiforme, Stevens-Johnson syndrome, sweat"**, toxic epidermal necrolysis (Lyell's syndrome).
Urogenital: acute interstitial nephritis, cystitis**, dysuria**, hematuria, increase in menstrual flow, nephrotic syndrome, oliguria/polyuria**, proteinuria**, renal insufficiency, acute renal failure, decreased menstrual flow.
OVERDOSAGE
First paragraph, last sentence added -
Anaphylactoid reactions have been reported with therapeutic ingestion of NSAIDs, and may occur following an overdose.
DOSAGE AND ADMINISTRATION
Individualization of dosage:
First paragraph, third sentence revised and fourth sentence added:
In osteoarthritis and rheumatoid arthritis and juvenile rheumatoid arthritis, the dosage
should be individualized to the lowest effective dose of Daypro to minimize adverse effects. The maximum recommended total daily dose of Daypro in adults is 1800 mg (26 mg/kg, whichever is lower) in divided doses. In children, doses greater than 1200 mg have not been studied.
Third paragraph, first sentence revised:
In adults, in cases where a quick onset of action is important, the pharmacokinetics of oxaprozin allow therapy to be started with a one-time loading dose of 1200 to 1800 mg (not to exceed 26 mg/kg).
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
DEPACON (valproate sodium) Injection
[January 24, 2002: Abbott]
CLINICAL PHARMACOLOGY
Pharmacokinetics
Bioavailability
Eleven healthy volunteers were given single infusions of 1000mg IV valproate over 5, 10, 30, and 60 minutes in a 4 -period crossover study. Total valproate concentrations were measured; unbound concentrations were not measured. After the 5 minute infusions (mean rate of 2.8 mg/kg/min), mean Cmax was 145 ± 32 µg/mL, while after the 60 minute infusions, mean Cmax was 115 ± 8 µg/mL. Ninety to 120 minutes after infusion initiation, total valproate concentrations were similar for all 4 rates of infusion. Because protein binding is nonlinear at higher total valproate concentrations, the corresponding increase in unbound Cmax at faster infusion rates will be greater.
ADVERSE EVENTS
In a separate clinical safety trial, 112 patients with epilepsy were given infusions of Depacon (up to 15mg/kg) over 5 to 10 minutes (1.5-3.0 mg/kg/min). The common adverse events (>2%) were somnolence (10.7%), dizziness (7.1%), paresthesia (7.1%), asthenia (7.1%), nausea (6.3%) and headache (2.7%). While the incidence of these adverse events was generally higher than in Table1 (experience encompassing the standard, much slower infusion rates), e.g. somnolence (1.7%), dizziness (5.2%), paresthesia (0.9%), asthenia (0%), nausea (3.2%), and headache (4.3%), a direct comparison between the incidence of adverse events in the 2 cohorts cannot be made because of differences in patient populations and study designs.
DOSAGE AND ADMINISTRATION
In one clinical safety study, approximately 90 patients with epilepsy and with no measurable plasma levels of valproate were given single infusions of Depacon (up to 15mg/kg and mean dose of 1184mg) over 5-10 minutes (1.5-3.0mg/kg/min). Patients generally tolerated the more rapid infusions well (see ADVERSE REACTIONS). This study was not designed to assess the effectiveness of these regimens. For pharmacokinetics with rapid infusions, see CLINICAL PHARMACOLOGY, Pharmacokinetics – Bioavailability.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
DILATRATE-SR (isosorbide dinitrate) Sustained Release Capsules
[January 30, 2002: Schwarz Pharma]
PRECAUTIONS
Geriatric Use:
Clinical studies of Dilatrate-SR did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
DIPENTUM (olsalazine sodium) Capsules
[January 18, 2002: Pharmacia & Upjohn]
PRECAUTIONS
Geriatric Use: Clinical studies of DIPENTUM did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patents.
In general, elderly patients should be treated with caution due to the greater frequency of decreased hepatic, renal, or cardiac function, co-existence of other disease, as well as concomitant drug therapy.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
DOXIL (doxorubicin HCl liposome) Injection
[January 10, 2002: ALZA]
Boxed WARNING
| 2. | Acute infusion-related |
WARNINGS
Cardiac Toxicity
End of the fourth paragraph, the following has been added:
(see ADVERSE REACTIONS; cardiac events)
Myelosuppression
Second sentence of the first paragraph revised:
From: Anemia was the most common hematologic adverse event (52.6%), followed by neutropenia (51.7%), leukopenia (WBC < 4000 mm3; 42.2%), and thrombocytopenia (24.2%).
To: Anemia was the most common hematologic adverse event (52.6%), followed by leukopenia (WBC < 4000 mm3; 42.2%), thrombocytopenia (24.2%), and neutropenia [ANC < 1000] (19.0%) (See Hemotology Data table in ADVERSE REACTIONS, Ovarian Cancer Patients.)
Infusion Reactions
Reactions were added to the first sentence of the first paragraph:
Acute infusion-related reactions, characterized by flushing, shortness of breath, facial swelling, headache, chills, chest pain, back pain, tightness in the chest and throat, fever, tachycardia, pruritis, rash, cyanosis, syncope, bronchospasm, asthma, apnea, and/or hypotension have occurred in 5% to 10% of patients treated with Doxil.
Second paragraph was added as follows:
Serious and sometimes life-threatening or fatal allergic/anaphylactoid-like infusion reactions have been reported. Medications to treat such reactions, as well as emergency equipment, should be available for immediate use.
ADVERSE REACTIONS
Ovarian Cancer Patients
Hematology Data Reported in Ovarian Cancer Patients table, the Leukopenia category was deleted and the Neutropenia category of < 2000 mm3 was replaced with a category of < 1000 mm3.
AIDS-KS Patients
Hematology Data Reported in AIDS-KS Patients table, the Leukopenia category
was deleted and the Neutropenia category of <
2000 mm3 was replaced with a category of <1000
mm3.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
GLUCOPHAGE XR & GLUCOPHAGE (metformin HCl) Tablets
[January 8, 2002: Bristol-Myers Squibb]
[Other labeling changes not appearing in 2001 PDR:
http://www.fda.gov/medwatch/SAFETY/2001/feb01.htm#glucop,
http://www.fda.gov/medwatch/safety/2000/dec00.htm#glucop]
PRECAUTIONS
Information for Patients
Patients should be informed that Glucophage XR must be swallowed whole and not crushed or chewed, and that the inactive ingredients may occasionally be eliminated in the feces as a soft mass that may resemble the original tablet.
DOSAGE AND ADMINISTRATION:
GLUCOPHAGE XR tablets must be swallowed whole and never crushed or chewed. Occasionally, the inactive ingredients of GLUCOPHAGE XR will be eliminated in the feces as a soft, hydrated mass. (See Patient Information Printed Below).
Patient Information page:
GLUCOPHAGE XR must be swallowed whole and never crushed or chewed. Occasionally, the inactive ingredients of GLUCOPHAGE XR may be eliminated as a soft mass in your stool that may look like the original tablet; this is not harmful and will not affect the way GLUCOPAHGE XR works to control your diabetes.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
Glofil-125 (sodium lothalamate) Injection
[January 22, 2002: Questcor Pharmaceuticals]
CONTRAINDICATIONS
Glofil-125 should not be administered via a central venous line.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
HYTRIN (terazosin HCl) Tablets & Soft Elastic Capsules
[January 23, 2002: Abbott]
ADVERSE REACTIONS
Post-marketing Experience
Post-marketing experience indicates that in rare instances patients may develop allergic reactions, including anaphylaxis, following administration of terazosin hydrochloride. There have been reports of priapism and thrombocytopenia during post-marketing surveillance. Atrial fibrillation has been reported.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
KALETRA (lopinavir/ritonavir) Capsules & Oral Solution
[January 18, 2002: Abbott]
Labeling contains 48-week safety and efficacy data (updated from 24-week data) from three studies included in the original NDA. These supplements provide for the use of KALETRA Capsules and KALETRA Oral Solution in combination with other antiretroviral agents for the treatment of HIV-infection. This indication is based on analyses of plasma HIV RNA levels and CD4 cell counts in a controlled study of KALETRA of 48 weeks duration and in smaller uncontrolled dose-ranging studies of KALETRA of 72 weeks duration. At present, there are no results from controlled trials evaluating the effect of KALETRA on clinical progression of HIV.
CLINICAL PHARMACOLOGY
Microbiology
Mechanism of action
Resistance: Phase III study of 653 (study 863)
Clinical Studies - Antiviral activity of KALETRA in patients with previous protease inhibitor therapy.therapies Study 957 Table 1 48 week response
Table 1: HIV RNA Response at Week 48 by baseline KALETRA susceptibility and by number of protease inhibitor-associated mutations1
For labeling changes found in CLINICAL PHARMACOLOGY subsection above, contact the company for the new label/package insert.
INDICATIONS AND USAGE
KALETRA is indicated in combination with other antiretroviral agents for the treatment of HIV-infection. This indication is based on analyses of plasma HIV RNA levels and CD4 cell counts in a controlled study of KALETRA of 242424 48 weeks duration and in smaller uncontrolled dose-ranging studies of KALETRA of 72 weeks duration. At present, there are no results from controlled trials evaluating the effect of KALETRA on clinical progression of HIV.
Figure 2: Virologic Response Through 24 WEEKS
(Study 863) 24 Weeks (Study 863)Week
48, Study 863*†
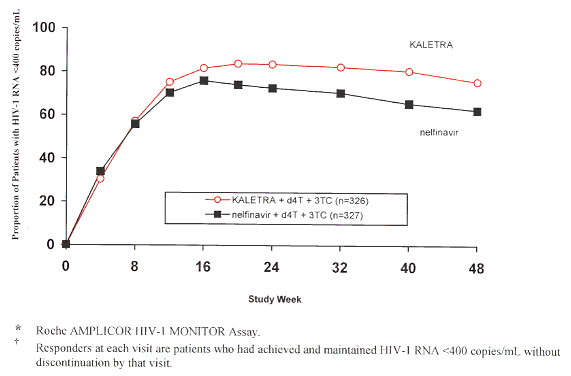
Table 3: 3:4: Outcomes of Randomized Treatment Through Week 24 - 24 -48 (Study 863)
|
Outcome |
KALETRA+d4T+3TC (N=326) |
Nelfinavir+d4T+3TC (N=327) |
|
Responder*1 |
75% |
62% |
|
Virologic failure2 Rebound Never suppressed through Week 48 |
9% 7% 2% |
25% 15% 9% |
|
Death |
2% |
1% |
|
Discontinued due to adverse event |
4% |
4% |
|
Discontinued for other reasons3 |
10% |
8% |
* Corresponds to rates at Week 48 in Figure 2. 1 Patients achieved and maintained confirmed HIV RNA <400 copies/mL through Week 48. 2 Includes confirmed viral rebound and failure to achieve confirmed <400 copies/mL through Week 48. 3 Includes lost to follow-up, patient’s withdrawal, non-compliance, protocol violation and other reasons. |
||
Through 48 weeks of therapy, there was a statistically significantly higher proportion of patients in the KALETRA arm compared to the nelfinavir arm with HIV RNA <400 copies/mL (75% vs. 62%, respectively) and HIV RNA <50 copies/mL (67% vs. 52%, respectively). Treatment response by baseline HIV RNA level subgroups is presented in Table 5.
Table 5: Proportion of Responders Through Week 48 by Baseline Viral Load (Study 863)
|
Baseline Viral Load (HIV-1 RNA copies/mL) |
KALETRA +d4T+3TC |
Nelfinavir +d4T+3TC |
||||
|
<400 copies/mL1 |
<50 copies/mL2 |
n |
<400 copies/mL1 |
<50 copies/mL2 |
n |
|
|
<30,000 |
74% |
71% |
82 |
79% |
72% |
87 |
|
≥30,000 to <100,000 |
81% |
73% |
79 |
67% |
54% |
79 |
|
≥100,000 to <250,000 |
75% |
64% |
83 |
60% |
47% |
72 |
|
≥250,000 |
72% |
60% |
82 |
44% |
33% |
89 |
1 Patients achieved and maintained confirmed HIV RNA <400 copies/mL through Week 48. |
||||||
2 Patients achieved HIV RNA <50 copies/mL at Week 48. |
||||||
Through 24Through 24
48 weeks of therapy, the mean increase from baseline in CD4 cell
count was 154 154207
cells/mm3 for the KALETRA arm and 150
150195 cells/mm3 for the nelfinavir
arm.
PRECAUTIONS
Hepatic Impairment and Toxicity
KALETRA is principally metabolized by the liver; therefore, caution should be exercised when administering this drug to patients with hepatic impairment, because lopinavir concentrations may be increased. Patients with underlying hepatitis B or C or marked elevations in transaminases prior to treatment may be at increased risk for developing further transaminase elevations.
elevations or hepatic decompensation. There have been postmarketing reports of hepatic dysfunction, including some fatalities. These have generally occurred in patients with advanced HIV disease taking multiple concomitant medications in the setting of underlying chronic hepatitis or cirrhosis. A causal relationship with KALETRA therapy has not been established. Increased AST/ALT monitoring should be considered in these patients, especially during the first several months of KALETRA treatment.
Fat Redistribution
Redistribution/accumulation of body fat including central obesity, dorsocervical fat enlargement (buffalo hump), peripheral wasting, facial wasting, breast enlargement, and "cushingoid appearance" have been observed in patients receiving antiretroviral therapy. The mechanism and long-term consequences of these events are currently unknown. A causal relationship has not been established.
Pediatric Use
Fourth paragraph -
Through 24 2448 weeks of therapy, the proportion of patients withwho achieved and sustained an HIV RNA < 400 copies/mL was 82 82%80% for antiretroviral naive patients and 66 66%71% for antiretroviral experienced patients. The mean increase from baseline in CD4 cell count was 328 328404 cells/mm3 for antiretroviral naive and 335335 284cells/mm3 for antiretroviral experienced patients treated through 24 24 weeks. At 24 weeks, one patient (1%)48 weeks. At 24 weeks, one patient (1%)At 48 weeks, two patients (2%) had prematurely discontinued the study. This discontinuation wasOne antiretroviral naive patient prematurely discontinued secondary to an adverse event attributed to KALETRA, while one antiretroviral experienced patient prematurely discontinued secondary to an HIV-relatedevent in an antiretroviral experienced patient that was not attributed to a KALETRA adverse event.
ADVERSE REACTIONS
Adults:
Treatment-Emergent Adverse Events: KALETRA has been studied in 612 patients as combination therapy in Phase I/II and Phase III clinical trials. The most common adverse event associated with KALETRA therapy was diarrhea, which was generally of mild to moderate severity. Rates of discontinuation of randomized therapy due to adverse events were 2.8% in KALETRA and 3.1% in nelfinavir treated 2.8% in KALETRA and 3.1% in nelfinavir treated5.8% in KALETRA-treated and 4.9% in nelfinavir-treated patients in Study 863.
Drug related clinical adverse events of moderate or severe intensity in > 2% of patients treated with combination therapy including KALETRA for up to 24 2448 weeks (Phase III) and for up to 72 weeks (Phase I/II) are presented in Table 7.9. For other information regarding observed or potentially serious adverse events, please see WARNINGS and PRECAUTIONS.
Table 7 7:9: Percentage of Patients with Treatment-Emergent1 Adverse Events of Moderate or Severe Intensity Reported in > 2% of Adult Patients
|
Antiretroviral Naive Patients |
Protease Inhibitor Experienced Patients |
|||
|
Study 863 (48 Weeks) |
Study 720 (72 Weeks) |
Phase I/II and Phase III |
||
|
KALETRA 400/100 mg BID + d4T + 3TC (N=326) |
Nelfinavir 750 mg TID + d4T + 3TC (N=327) |
KALETRA BID2 + d4T + 3TC (N= 84) |
KALETRA BID3 + NNRTI + NRTIs (N= 186) |
|
|
Body as a Whole |
||||
|
Abdominal Pain |
4.0% |
3.1% |
4.8% |
1.6% |
|
Asthenia |
4.0% |
3.4% |
7.1% |
5.4% |
|
Headache |
2.5% |
1.8% |
7.1% |
1.6% |
|
Pain |
0.6% |
0.0% |
2.4% |
1.6% |
|
Digestive System |
||||
|
Abnormal Stools |
0.0% |
0.3% |
6.0% |
1.6% |
|
Diarrhea |
15.6% |
17.1% |
23.8% |
15.6% |
|
Dyspepsia |
2.1% |
0.3% |
1.2% |
0.5% |
|
Nausea |
6.7% |
4.6% |
15.5% |
2.7% |
|
Vomiting |
2.5% |
2.4% |
4.8% |
1.6% |
|
Nervous System |
||||
|
Insomnia |
1.5% |
1.2% |
2.4% |
1.1% |
|
Skin and Appendages |
||||
|
Rash |
0.6% |
1.5% |
3.6% |
2.0% |
Treatment-emergent adverse events occurring in less than 2% of adult patients receiving KALETRA in all phase II/III clinical trials and considered at least possibly related or of unknown relationship to treatment with KALETRA and of at least moderate intensity are listed below by body system.
Body as a Whole: Back BackAbdomen enlarged, back pain, chest pain, chest pain substernal, chills, cyst, drug interaction, drug level increased, face edema, fever, flu syndrome, hypertrophy, infection bacterial, malaise, and viral infection.
Cardiovascular System: Deep vein thrombosis, hypertension, migraine, palpitation, thrombophlebitis, varicose vein, and vasculitis.
Digestive System: Anorexia, cholecystitis, constipation, dry mouth,dyspepsia, dyspepsia, dysphagia, enterocolitis, eructation, esophagitis, fecal incontinence, flatulence, gastritis, gastroenteritis, gastrointestinal disorder, hemorrhagic colitis, increased appetite, mouth ulceration, pancreatitis, sialadenitis, stomatitis, and ulcerative stomatitis.
Endocrine System: Cushing’s syndrome, diabetes mellitus, and hypothyroidism.
Hemic and Lymphatic System: Anemia, leukopenia, and lymphadenopathy.
Metabolic and Nutritional Disorders: Avitaminosis, dehydration, edema, glucose tolerance decreased, lactic acidosis, obesity, peripheral edema, weight gain, and weight loss.
Musculoskeletal System: Arthralgia, arthrosis and myalgia.
Nervous System: Abnormal dreams, agitation, amnesia, anxiety, ataxia, confusion, depression, dizziness, dyskinesia, emotional lability, encephalopathy, facial paralysis, hypertonia, libido decreased, nervousness, neuropathy, paresthesia, peripheral neuritis, somnolence, thinking abnormal, and tremor.
Respiratory System: Bronchitis, dyspnea, lung edema, rhinitis, and sinusitis.
Skin and Appendages: Acne, alopecia, dry skin, eczema, exfoliative dermatitis, furunculosis, maculopapular rash, nail disorder, pruritis, seborrhea, skin benign neoplasm, skin discoloration, skin ulcer, and sweating.
Special Senses: Abnormal vision, eye disorder, otitis media, taste perversion, and tinnitus.
Urogenital System: Abnormal ejaculation, gynecomastia, hypogonadism male, kidney calculus, and urine abnormality.
Post-Marketing Experience: Redistribution/accumulation of body fat has been reported (see PRECAUTIONS, Fat Redistribution).
Table 8 8:10: Grade 3-4 Laboratory Abnormalities Reported in > 2% of Adult Patients
|
Variable |
Limit1 |
Antiretroviral Naive Patients |
Antiretroviral Experienced Patients |
||
|
Study 863 (48 Weeks) |
Study 720 (72 Weeks) |
Phase I/II and Phase III |
|||
|
KALETRA 400/100 mg BID + d4T + 3TC (N=326) |
Nelfinavir 750 mg TID + d4T + 3TC (N=327) |
KALETRA BID2 + d4T + 3TC (N=84) |
KALETRA BID3 + NNRTI + NRTIs (N=186) |
||
|
Chemistry |
High |
||||
|
Glucose |
>250 mg/dL |
2.2% |
1.6% |
2.4% |
4.4% |
|
Uric Acid |
>12 mg/dL |
2.2% |
1.6% |
3.6% |
0.5% |
|
SGOT/AST |
>180 U/L |
2.2% |
4.1% |
9.5% |
5.5% |
|
SGPT/ALT |
>215 U/L |
3.8% |
3.8% |
8.3% |
7.1% |
|
GGT |
>300 U/L |
N/A |
N/A |
3.6% |
24.6%4 |
|
Total Cholesterol |
>300 mg/dL |
9.0% |
5.0% |
14.3% |
28.4% |
|
Triglycerides |
>750 mg/dL |
9.3% |
1.3% |
10.7% |
28.4% |
|
Amylase |
>2 x ULN |
3.2% |
2.2% |
4.8% |
4.9% |
|
Chemistry |
Low |
||||
|
Inorganic Phosphorus |
<1.5 mg/dL |
0.0% |
0.0% |
0.0% |
2.2% |
|
Hematology |
Low |
||||
|
Neutrophils |
0.75 x 109/L |
0.6% |
2.5% |
2.4% |
2.7% |
Pediatrics:
Treatment-Emergent Adverse Events: KALETRA has been studied in 100 pediatric patients 6 months to 12 years of age. The adverse event profile seen during a clinical trial was similar to that for adult patients.
Taste aversion, vomiting, and diarrhea were the most commonly reported drug related adverse events of any severity in pediatric patients treated with combination therapy including KALETRA for up to 48 weeks in Study 940. A total of 8 children experienced moderate or severe adverse events at least possibly related to Rash (2%)KALETRA. Rash (reported in 3%) was the only drug-related clinical adverse events of moderate orto severe intensity in > 2% of pediatric patients treated with combination therapy including KALETRA (300/75 mg/m2) for up to 24 weeks (Study 940). This includes adverse events of at least possible, probable or unknown relationship to study drug.observed in ³ 2% of children enrolled.
Table 9 9:11: Grade 3-4 Laboratory Abnormalities Reported in ³
2% Pediatric Patients
|
Variable |
Limit1 |
KALETRA BID+ RTIs (N=100) |
|
Chemistry |
High |
|
|
Sodium |
> 149 mEq/L |
3.0% |
|
Total bilirubin |
³ 3.0 x ULN |
3.0% |
|
SGOT/AST |
> 180 U/L |
8.0% |
|
SGPT/ALT |
> 215 U/L |
7.0% |
|
Total cholesterol |
> 300 mg/dL |
3.0% |
|
Amylase |
> 2.5 x ULN |
7.0%2 |
|
Chemistry |
Low |
|
|
Sodium |
< 130 mEq/L |
3.0% |
|
Hematology |
Low |
|
|
Platelet Count |
< 50 x 109/L |
4.0% |
|
Neutrophils |
< 0.40 x 109/L |
2.0% |
2 Subjects with Grade 3-4 amylase confirmed by elevations in pancreatic amylase.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
Lidocaine HCl in 5% Dextrose Injection
[January 23, 2002: Baxter Healthcare]
Precautions
Drug Interactions - "amiodarone" has been added to the first sentence of the third paragraph.
Geriatric Use -
Clinical studies of Lidocaine Hydrochloride did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
LOVENOX (enoxaparin sodium) Injection
[January 9, 2002: Aventis]
WARNINGS
Prosthetic Heart Valves: The use of Lovenox Injection is not recommended for thromboprophylaxis in patients with prosthetic heart valves. Cases of prosthetic heart valve thrombosis have been reported in patients with prosthetic valves who have received enoxaparin for thromboprophylaxis. Some of these cases were pregnant women in whom thrombosis led to maternal deaths and fetal deaths. Pregnant women with prosthetic heart valves may be at higher risk for thromboembolism (see PRECAUTIONS: Pregnancy).
PRECAUTIONS
Pregnancy
Teratogenic Effects
Second paragraph added:
There have been reports of congenital anomalies in infants born to women who received enoxaparin during pregnancy including cerebral anomalies, limb anomalies, hypospadias, peripheral vascular malformation, fibrotic dysplasia, and cardiac defect. A cause and effect relationship has not been established nor has the incidence been shown to be higher than in the general population.
Non-Teratogenic Effects
First paragraph revised:
Non-teratogenic Effects: There have been a few spontaneous post-marketing reports of fetal death when pregnant women received enoxaparin. Causality of the cases has not been determined. In one case, placental hemorrhage and detachment were found in association with the fetal death. If enoxaparin is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to the fetus.
There have been post-marketing reports of fetal death when pregnant women received Lovenox Injection. Causality for these cases has not been determined. Pregnant women receiving anti-coagulants, including enoxaparin, are at increased risk for bleeding. Hemorrhage can occur at any site and may lead to death of mother and/or fetus. Pregnant women receiving enoxaparin should be carefully monitored. Pregnant women and women of child-bearing potential should be apprised of the potential hazard to the fetus and the mother if enoxaparin is administered during pregnancy.
Second paragraph added:
In a clinical study of pregnant women with prosthetic heart valves given enoxaparin (1 mg/kg bid) to reduce the risk of thromboembolism, 2 of 7 women developed clots resulting in blockage of the valve and leading to maternal and fetal death. There are postmarketing reports of prosthetic valve thrombosis in pregnant women with prosthetic heart valves while receiving enoxaparin for thromboprophylaxis. These events resulted in maternal death or surgical interventions. The use of Lovenox Injection is not recommended for thromboprophylaxis in pregnant women with prosthetic heart valves (see WARNINGS: Prosthetic Heart Valves).
ADVERSE REACTIONS
Ongoing Safety Surveillance: Since 1993, there have been over 79 reports 80 reports of epidural or spinal hematoma formation with concurrent use of Lovenox Injection and spinal/epidural anesthesia or spinal puncture.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
MINOCIN (minocycline HCl) I.V.
[January 30, 2002: WYETH-AYERST RESEARCH
INDICATION AND USAGE:
Addition of "Nongonococcal urethritis, endocervical, or rectal infections in adults caused by Ureaplasma urealyticum or Chlamydia trachomatis."
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
NORPLANT SYSTEM (levonorgestrel implant)
[January 15: 2002: Wyeth Ayerst]
WARNINGS
B. Warnings Based on Experience with Combination (Progestin plus Estrogen) Oral Contraceptives
3.Carcinoma
Previous subsection replaced with the following:
A meta-analysis from 54 epidemiological studies reported that there is a slightly increased relative risk (RR=1.24) of having breast cancer diagnosed in women who are currently using combination oral contraceptives compared to never-users. The increased risk gradually disappears during the course of the 10 years after cessation of combination oral contraceptive use. These studies do not provide evidence for causation. The observed pattern of increased risk of breast cancer diagnosis may be due to earlier detection of breast cancer in combination oral contraceptive users, the biological effects of combination oral contraceptives, or a combination of both. Because breast cancer is rare in women under 40 years of age, the excess number of breast cancer diagnoses in current and recent combination oral contraceptive users is small in relation to the lifetime risk of breast cancer. Breast cancers diagnosed in ever-users tend to be less advanced clinically than the cancers diagnosed in never-users. Although the results were broadly similar for progestin-only oral contraceptives, the data are based on much smaller numbers of progestin-only oral contraceptive users and therefore are less conclusive than for combination oral contraceptives. This information should be considered when prescribing the NORPLANT SYSTEM.
PATIENT LABELING
Risks of Using the Norplant System section - Contact the company for a copy of the labeling/package insert.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
OXYCONTIN (oxycodone HCl) Controlled Release Tablets
[January 15, 2002: Purdue Pharma]
[Other safety related information regarding Oxycontin: http://www.fda.gov/medwatch/safety/2001/safety01.htm#oxycon]
Patient Information sheet added:
PATIENT INFORMATION
OXYCONTIN Schedule II
(Oxycodone HCl Controlled-Release) Tablets
OxyContin Tablets, 10 mg
OxyContin Tablets, 20 mg
OxyContin Tablets, 40 mg
OxyContin Tablets, 80 mg
OxyContin Tablets, 160 mg
Read this information carefully before you take OxyContin (ox-e-CON-tin) tablets. Also read the information you get with your refills. There may be something new. This information does not take the place of talking with your doctor about your medical condition or your treatment. Only you and your doctor can decide if OxyContin is right for you. Share the important information in this leaflet with members of your household.
What Is The Most Important Information I Should Know About OxyContin ?
• Use OxyContin the way your doctor tells you to.
• Use OxyContin only for the condition for which it was prescribed.
• OxyContin is not for occasional ("as needed") use.
• Swallow the tablets whole. Do not break, crush, dissolve, or chew them before swallowing.
OxyContin works properly over 12 hours only when swallowed whole. If a tablet is broken, crushed, dissolved, or chewed, the entire 12 hour dose will be absorbed into your body all at once. This can be dangerous, causing an overdose, and possibly death.
• Keep OxyContin out of the reach of children. Accidental overdose by a child is dangerous and may result in death.
• Prevent theft and misuse. OxyContin contains a narcotic painkiller that can be a target for people who abuse prescription medicines. Therefore, keep your tablets in a secure place, to protect them from theft. Never give them to anyone else . Selling or giving away this medicine is dangerous and against the law.
What is OxyContin ?
OxyContin is a tablet that comes in several strengths and contains the medicine oxycodone (ox-e-KOE- done). This medicine is a painkiller like morphine. OxyContin treats moderate to severe pain that is expected to last for an extended period of time. Use OxyContin regularly during treatment. It contains enough medicine to last for up to twelve hours.
Who Should Not Take OxyContin ?
Do not take OxyContin if
· your doctor did not prescribe OxyContin for you.
· your pain is mild or will go away in a few days.
· your pain can be controlled by occasional use of other painkillers.
· you have severe asthma or severe lung problems.
· you have had a severe allergic reaction to codeine, hydrocodone, dihydrocodeine, or oxycodone (such as Tylox, Tylenol with Codeine, or Vicodin. A severe allergic reaction includes a severe rash, hives, breathing problems, or dizziness.
· you had surgery less than 12 - 24 hours ago and you were not taking OxyContin just before surgery.
Your doctor should know about all your medical conditions before deciding if OxyContin is right for you and what dose is best. Tell your doctor about all of your medical problems, especially the ones listed below:
· trouble breathing or lung problems
· head injury
· liver or kidney problems
· adrenal gland problems, such as Addison’s disease
· convulsions or seizures
· alcoholism
· hallucinations or other severe mental problems
· past or present substance abuse or drug addiction
If any of these conditions apply to you, and you haven’t told your doctor, then you should tell your doctor before taking OxyContin.
If you are pregnant or plan to become pregnant, talk with your doctor. OxyContin may not be right for you. Tell your doctor if you are breast feeding. OxyContin will pass through the milk and may harm the baby.
Tell your doctor about all the medicines you take, including prescription and non-prescription medicines, vitamins, and herbal supplements. They may cause serious medical problems when taken with OxyContin, especially if they cause drowsiness.
How Should I Take OxyContin ?
· Follow your doctor’s directions exactly. Your doctor may change your dose based on your reactions to the medicine. Do not change your dose unless your doctor tells you to change it. Do not take OxyContin more often than prescribed.
· Swallow the tablets whole. Do not break, crush, dissolve, or chew before swallowing. If the tablets are not whole, your body will absorb too much medicine at one time. This can lead to serious problems, including overdose and death.
· If you miss a dose, take it as soon as possible. If it is almost time for your next dose, skip the missed dose and go back to your regular dosing schedule. Do not take 2 doses at once unless your doctor tells you to.
· In case of overdose, call your local emergency number or poison control center right away.
· Review your pain regularly with your doctor to determine if you still need OxyContin.
· You may see tablets in your stools (bowel movements). Do not be concerned. Your body has already absorbed the medicine.
If you continue to have pain or bothersome side effects, call your doctor.
Stopping OxyContin. Consult your doctor for instructions on how to stop this medicine slowly to avoid uncomfortable symptoms. You should not stop taking OxyContin all at once if you have been taking it for more than a few days.
After you stop taking OxyContin, flush the unused tablets down the toilet.
What Should I Avoid While Take OxyContin ?
· Do not drive, operate heavy machinery, or participate in any other possibly dangerous activities until you know how you react to this medicine. OxyContin can make you sleepy.
· Do not drink alcohol while using OxyContin. It may increase the chance of getting dangerous side effects.
· Do not take other medicines without your doctor’s approval. Other medicines include prescription and non-prescription medicines, vitamins, and supplements. Be especially careful about products that make you sleepy.
What are the Possible Side Effects of OxyContin?
Call your doctor or get medical help right away if
• your breathing slows down
• you feel faint, dizzy, confused, or have any other unusual symptoms
Some of the common side effects of OxyContin are nausea, vomiting, dizziness, drowsiness, constipation, itching, dry mouth, sweating, weakness, and headache. Some of these side effects may decrease with continued use.
There is a risk of abuse or addiction with narcotic painkillers. If you have abused drugs in the past, you may have a higher chance of developing abuse or addiction again while using OxyContin. We do not know how often patients with continuing (chronic) pain become addicted to narcotics, but the risk has been reported to be small.
These are not all the possible side effects of OxyContin. For a complete list, ask your doctor or pharmacist.
General Advice About OxyContin
• Do not use OxyContin for conditions for which it was not prescribed.
• Do not give OxyContin to other people, even if they have the same symptoms you have. Sharing is illegal and may cause severe medical problems, including death.
This leaflet summarizes the most important information about OxyContin. If you would like more information, talk with your doctor. Also, you can ask your pharmacist or doctor for information about OxyContin that is written for health professionals.
CAUTION: Federal law prohibits dispensing without prescription.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
Plavix (clopidogrel bisulfate) Tablets
[January 28, 2002:Sanofi-Synthelabo]
ADVERSE REACTIONS
Postmarketing Experience
Added: fever
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
PRANDIN (repaglinide) Tablets
[January 18, 2002: Novo Nordisk Pharmaceuticals]
CLINICAL PHARMACOLOGY
Drug-Drug Interactions:
Drug interaction studies performed in healthy volunteers show that PRANDIN had no clinically relevant effect on the pharmacokinetic properties of digoxin, theophylline, or warfarin. Co-administration of cimetidine with PRANDIN did not significantly alter the absorption and disposition of repaglinide.
Additionally, the following drugs were studied in healthy volunteers with co-administration of PRANDIN. Listed below are the results:
Ketoconazole: Co-administration of 200 mg ketoconazole and a single dose of 2 mg PRANDIN (after 4 days of once daily ketoconazole 200 mg) resulted in a 15% and 16% increase in repaglinide AUC and Cmax, respectively. The increases were from 20.2 ng/ml to 23.5 ng/ml for Cmax and from 38.9 ng/mL *hr to 44.9 ng/mL *hr for AUC.
Rifampin: Co-administration of 600 mg rifampin and a single dose of 4 mg Prandin (after 6 days of once daily rifampin 600 mg) resulted in a 32% and 26% decrease in repaglinide AUC and Cmax, respectively. The decreases were from 40.4 ng/ml to 29.7 ng/ml for Cmax and from 56.8 ng/mL *hr to 38.7 ng/mL *hr for AUC.
Levonorgestrel & Ethinyl Estradiol: Co-administration of a combination tablet of 0.15 mg levonorgestrel and 0.03 mg ethinyl estradiol administered once daily for 21 days with 2 mg PRANDIN administered three times daily (days 1-4) and a single dose on Day 5 resulted in 20% increases in repaglinide, levonorgestrel, and ethinyl estradiol Cmax. The increase in repaglinide Cmax was from 40.5 ng/ml to 47.4 ng/ml. Ethinyl estradiol AUC parameters were increased by 20%, while repaglinide and levonorgestrel AUC values remained unchanged.
Simvastatin: Co-administration of 20 mg simvastatin and a single dose of 2 mg PRANDIN (after 4 days of once daily simvastatin 20 mg and three times daily Prandin 2 mg) resulted in a 26% increase in repaglinide Cmax from 23.6 ng/ml to 29.7 ng/ml. AUC was unchanged.
Nifedipine: Co-administration of 10 mg nifedipine with a single dose of 2 mg PRANDIN (after 4 days of three times daily nifedipine 10 mg and three times daily PRANDIN 2 mg) resulted in unchanged AUC and Cmax values for both drugs.
Special Populations
Renal insufficiency
Measures of AUC and Cmax after multiple dosing
of 2 mg repaglinide were found to be higher in three groups of patients with
reduced renal function: (AUCrmild/moderate impairment: 90.8 ng/mL*hr to AUCsevere
impairment: 137.7 ng/mL*hr versus AUChealthy:29.1 ng/mL*hr; Cmax, mild/moderate
impairment: 46.7 ng/mL to Cmax, severe impairment: 44.0 ng/mL versus Cmax, healthy
:20.6 ng/mL). Repaglinide AUC
is only weakly correlated to creatinine clearance. Initial dosage adjustment
does not appear to be necessary, but Subsequent increases in PRANDIN should
be made carefully in patients with type 2 diabetes who have renal function impairment
or renal failure requiring hemodialysis.
Single-dose and steady-state pharmacokinetics of repaglinide were compared between patients with type 2 diabetes and normal renal function (CrCl > 80 mg/dL), mild to moderate renal function impairment (CrCl = 40 – 80 mg/dL), and severe renal function impairment (CrCl = 20 – 40 mg/dL). Both AUC and Cmax of repaglinide were similar in patients with normal and mild to moderately impaired renal function (mean values 56.7 ng/mL *hr vs 57.2 ng/mL *hr and 37.5 ng/mL vs 37.7 ng/mL, respectively.) Patients with severely reduced renal function had elevated mean AUC and Cmax values (98.0 ng/mL *hr and 50.7 ng/mL, respectively), but this study showed only a weak correlation between repaglinide levels and creatinine clearance. Initial dose adjustment does not appear to be necessary for patients with mild to moderate renal dysfunction.
However, patients with type 2 diabetes who have severe renal function impairment should initiate PRANDIN therapy with the 0.5 mg dose – subsequently, patients should be carefully titrated. Studies were not conducted in patients with creatinine clearances below 20 mg/mL or patients with renal failure requiring hemodialysis.
Hepatic insufficiency. A single-dose, open-label study was conducted in 12 healthy subjects and 12 patients with chronic liver disease (CLD) classified by Child-Pugh scale and caffeine clearance.
INDICATIONS AND USAGE
Last paragraph revised:
The Diabetes Control and Complications Trial (DCCT) demonstrated, in patients with type 1 diabetes, that improved glycemic control, as reflected by HbA1C and fasting glucose levels, was associated with a reduction in the diabetic complications retinopathy, neuropathy, and nephropathy. In considering the use of PRANDIN or other antidiabetic therapies, it should be recognized that controlling the blood glucose in type 2 diabetes has not been established to be effective in preventing the long-term cardiovascular and neural complications of diabetes. It has not been shown that the implications of the DCCT results also apply to patients with type 2 diabetes. Nonetheless, improved glycemic control appears to be an important goal in many patients with non-insulin-dependent disease because it is presumed that the mechanisms by which glucose causes complications is the same in both forms of diabetes.
In considering the use of PRANDIN or other antidiabetic therapies, it should be recognized that blood glucose control in type 2 diabetes has not been definitely established to be effective in preventing the long-term cardiovascular complications of diabetes. However, in patients with Type 1 diabetes, the Diabetes Control and Complications Trial (DCCT) demonstrated that improved glycemic control, as reflected by HbA1C and fasting glucose levels, was associated with a reduction in the diabetic complications retinopathy, neuropathy, and nephropathy.
4. Revisions to the PRECAUTIONS section, Drug-Drug Interactions subsection.
5. Revision to the ADVERSE REACTIONS section, Infrequent Adverse Events subsection, to include several rare, spontaneously reported post-marketing adverse events.
PRECAUTIONS
Drug Interactions
In vitro data indicate that repaglinide metabolism may be inhibited
by antifungal agents like ketoconazole and miconazole, and antibacterial agents
like erythromycin (cytochrome P450 3A4 inhibitors).
Drugs that induce the cytochrome P450 enzyme system 3A4 may increase repaglinide
metabolism; such drugs include troglitazone,
rifampin, barbiturates, and carbamazepine. No
systematically acquired data are available on increased or decreased plasma
levels with 3A4 inhibitors or inducers See
CLINICAL PHARMACOLOGY section,
Drug-Drug Interactions.
5. Revision to the ADVERSE REACTIONS section, Infrequent Adverse Events subsection, to include several rare, spontaneously reported post-marketing adverse events.
ADVERSE EVENTS
Infrequent adverse events (<1% of patients)
Although no causal relationship has been established, postmarketing experience includes reports of the following rare adverse events: alopecia, hemolytic anemia, pancreatitis, Stevens-Johnson Syndrome, and severe hepatic dysfunction.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
RITALIN & RITALIN-SR (methylphenidate HCl) Tablets
[January 11, 2002: Novartis]
CONTRAINDICATIONS
Ritalin is contraindicated during treatment with monoamine oxidase inhibitors, and also within a minimum of 14 days following discontinuation of a monoamine oxidase inhibitor (hypertensive crises may result).
PRECAUTIONS
Drug Interactions
Serious adverse events have been reported in concomitant use with clonidine, although no causality for the combination has been established. The safety of using methylphenidate in combination with clonidine or other centrally acting alpha-2 agonists has not been systemically evaluated.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
[January 8, 2002: Bristol-Myers Squibb]
Labeling provides for revisions to the prescriber labeling regarding Serzone and hepatic failure to incorporate a patient package insert and a "Dear Healthcare Practitioner" letter. Use the links below to see complete revised prescribing information.
"Dear Healthcare Practitioner" letter: http://www.fda.gov/medwatch/SAFETY/2002/serzone_deardoc.PDF
Full Revised Serzone Label: http://www.fda.gov/medwatch/SAFETY/2002/serzone_label.pdf
Patient Package Insert: http://www.fda.gov/medwatch/SAFETY/2002/serzone_PPI.pdf
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
STADOL NS (butorphanol tartrate) Nasal Spray
[January 14, 2002: Bristol-Myers Squibb]
[Other labeling changes not appearing in 2001 PDR:
http://www.fda.gov/medwatch/SAFETY/2001/jan01.htm#stadol]
PRECAUTIONS
Drug Interactions
The safety of using STADOL NS and IMITREX (sumatriptan) Nasal Spray during the same episode of migraine has not been established. However, it should be noted that both products are capable of producing transient increases in blood pressure.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
TEV-TROPIN (somatropin) Injection
[January 4, 2002: Bio-Technology General]
Labeling provides for a name change from Bio-Tropin to Tev-Tropin.
DOSAGE AND ADMINISTRATION:
Subcutaneous injection of greater than 1 mL of reconstituted solution is not Recommended.
PRECAUTIONS
Geriatric Use:
The safety and effectiveness of Tev-Tropin in patients aged 65 and over has not been evaluated in clinical studies. Elderly patients may be more sensitive to the action of Tev-Tropinand may be more prone to develop adverse reactions.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
VIAGRA (sildenafil citrate) Tablets
[January 2, 2002: Pfizer]
PRECAUTIONS
Drug Interactions
Effects of Other Drugs on VIAGRA
In Vivo Studies: (paragraph 4)
In healthy male volunteers, there was no evidence of a clinically significant effect of azithromycin (500 mg daily for 3 days) on the systemic exposure of sildenafil or its major circulating metabolite.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
VIDEX EC (didanosine) Delayed Release Capsules
[January 29, 2002: Bristol-Myers Squibb]
[Other labeling changes not appearing in 2001 PDR:
http://www.fda.gov/medwatch/SAFETY/2001/nov01.htm#videx]
Labeling provides for the inclusion of safety and efficacy data from the final results of study AI454-152 in the VIDEX EC (didanosine) Delayed Release Capsules package insert. Contact the company for a copy of the labeling/package insert.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
XOPENEX (levalbuterol HCl) Inhalation Solution
[January 30, 2002: Sepracor]
Labeling provides for the use of Xopenex (levalbuterol inhalation solution) for the treatment or prevention of bronchospasm in children 6 years of age and older with reversible obstructive airway disease. Contact the company for a copy of the labeling/package insert.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
ZANTAC (ranitidine HCl) Tablets, Syrup, GELdose Capsules &
EFFERdose Tablets, Granules, Injection & Injection Premixed
[January 7, 2002: GlaxoSmithKline]
[Other labeling changes not appearing in 2001 PDR: http://www.fda.gov/medwatch/SAFETY/2001/may01.htm#zantac]
ADVERSE REACTIONS
Integumentary: Rash, including rare cases of erythema multiforme. Rare cases of alopecia and vasculitis.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
ZIAGEN (abacavir sulfate) Tablets & Oral Solution
[January 28, 2002: GlaxoSmithKline]
PRECAUTIONS:
Carcinogenesis,Mutagenesis, and Impairment of Fertility:
Carcinogenesis, Mutagenesis, and Impairment of Fertility: Abacavir was administered orally at 3 dosage levels to separate groups of mice (60 females and 60 males per group) and rats (56 females and 56 males in each group) in carcinogenicity studies. Single doses were 55, 110, and 330 mg/kg per day in mice and 30, 120, and 600 mg/kg per day in rats. Results showered an increase in the incidence of malignant and non-malignant tumors. Malignant tumors occurred in the preputial gland of males and the clitoral gland of females in both species, and in the liver and thyroid gland in female rats. These observations were made at systemic exposures in the range of 6 to 32 times the human exposure at the recommended dose (300 mg twice daily). It is not known how predictive the results of rodent carcinogenicity studies may be for humans.
HOW SUPPLIED
ANIMAL TOXICOLOGY:
Myocardial degeneration was found in mice and rats following administration of abacavir for 2 years. The systemic exposures were equivalent to 7 to 24 times the expected systemic exposure in humans. The clinical relevance of this finding has not been determined.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
[January 29, 2002: GlaxoSmithKline]
[Other labeling changes not appearing in 2001 PDR:
CLINICAL PHARMACOLOGY
Special Populations
Geriatrics: Acyclovir plasma concentrations are higher in geriatric patients compared to younger adults, in part due to age-related changes in renal function. Dosage reduction may be required in geriatric patients with underlying renal impairment (see PRECAUTIONS: Geriatric Use).
PRECAUTIONS
Carcinogenesis, Mutagenesis, Impairment of Fertility
[Paragraghs three through five revised (most or all of paragraphs three and four deleted and sentences one through three of paragraph five deleted)]
Acyclovir was tested in 16 in vitro and in vivo genetic toxicity assays. Acyclovir was positive in 5 of the assays. Acyclovir did not impair fertility or reproduction in mice (450 mg/kg per day, PO) or in rats (25 mg/kg per day, SC). In the mouse study, plasma levels were the same as human levels, while in the rat study, they were 1 to 2 times human levels. At higher doses (50 mg/kg per day, SC) in rats and rabbits (1 to 2 and 1 to 3 times human levels, respectively) implantation efficacy, but not litter size, was decreased. In a rat peri- and post-natal study at 50 mg/kg per day, SC, there was a statistically significant decrease in group mean numbers of corpora lutea, total implantation sites, and live fetuses.
Pregnancy: Teratogenic Effects:
First paragraph:
Pregnancy Category B. Acyclovir administered during organogenesis was not teratogenic in the mouse (450 mg/kg per day, PO), rabbit (50 mg/kg per day, SC and IV), or rat (50 mg/kg per day, SC).
Second paragraph:
There are no adequate and well-controlled studies in pregnant women. A prospective
epidemiologic registry of acyclovir use during pregnancy has
collected data since June 1984. As of December 1997, outcomes of live births
have been documented in 552 women has
collected data since June 1984. As of December 1997, outcomes of live births
have been documented in 552 women was
established in 1984 and completed in April 1999. There were 749 pregnancies
followed in women exposed to systemic acyclovir during the first
trimester of pregnancy resulting in pregnancy.756
outcomes. The occurrence rate of birth defects approximates
that found in the general population. However, the small size of the registry
is insufficient to evaluate the risk for specific
specific less
common defects or to permit reliable or
definitive conclusions regarding the safety of acyclovir in pregnant women and
their developing fetuses. Acyclovir should
be used during pregnancy only if the potential benefit justifies the potential
risk to the fetus.
Geriatric Use: Clinical studies of
ZOVIRAX did not include sufficient numbers of patients aged 65 and over to determine
whether they respond differently than younger patients. In general, dose selection
for an elderly patient should be cautious, usually starting at the low end of
the dosing range, reflecting the greater frequency of decreased renal function,
and of concomitant disease or other drug therapy.
Geriatric Use: Clinical studies of ZOVIRAX for Injection did not include sufficient numbers of patients aged 65 and over to determine whether they respond differently from younger patients. Other reported clinical experience has identified differences in the severity of CNS adverse events between elderly and younger patients (see ADVERSE REACTIONS: Observed During Clinical Practice). In general, dose selection for an elderly patient should be cautious, reflecting the greater frequency of decreased renal function, and of concomitant disease or other drug therapy. This drug is known to be substantially excreted by the kidney, and the risk of toxic reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection, and it may be useful to monitor renal function.
ADVERSE REACTIONS
Observed During Clinical Practice
General: angioedema
Digestive: Diarrhea, elevated liver function
tests, elevated
liver function tests, gastrointestinal distress, nausea
Hematologic and Lymphatic: Disseminated intravascular coagulation, hemolysis, leukocytoclastic vasculitis, leukopenia, lymphadenopathy.
Hepatobiliary Tract and Pancreas: Elevated liver function tests, hepatitis, hyperbilirubinemia, jaundice.
Nervous: Aggressive behavior, agitation, ataxia, coma, confusion, delirium, dizziness, encephalopathy, hallucinations, obtundation, paresthesia, psychosis, seizure, somnolence, tremor. These symptoms may be marked, particularly in older adults (see PRECAUTIONS).
Skin: Alopecia, erythema multiforme, photosensitive rash, pruritus, rash, Stevens-Johnson syndrome, toxic epidermal necrolysis, urticaria. Severe local inflammatory reactions, including tissue necrosis, have occurred following infusion of ZOVIRAX into extravascular tissues.
HTML by JLW