Data and Safety Monitoring Policy

National Heart, Lung, and Blood Institute
National Institutes of Health
October 31, 2008
Purpose and Scope:
This policy sets forth the National Heart, Lung, and Blood
Institute (NHLBI) requirements for data and safety monitoring
(DSM) for all human
subjects research funded in whole or in part by NHLBI
extramural programs. Effective October 1, 2008, this policy
is incorporated as part of the terms and conditions for all
awards.
Release of funds for human subjects research activities is
contingent upon compliance with this DSM policy.
Policy:
- Data and safety monitoring is a critical component of
all human subjects research; the level of monitoring should
be commensurate with the complexity of the study and the
level of risk to study participants.
- All human subjects research requires Institutional Review
Board (IRB) approval. IRBs have the prime responsibility
for oversight of all clinical research conducted at their
institutions.
- Certification of IRB approval of the protocol must be
sent (preferably electronically) to NHLBI before a proposed
human subjects research project may begin and should include
the following elements:
- Application grant
number or contract
number
- Title of research project
- Principal investigator (PI) name
- Date of IRB approval or exemption
- Appropriate signatures (e.g., IRB chair, Authorized
Organizational Representative)
- For studies with multiple protocols, the IRB identification
number and approval date associated with each protocol
NOTE:
The awardee is responsible
for ensuring that all of its sites engaged in research involving
human subjects have IRB approval and an appropriate assurance
approved by the Office of Human Research Protections (OHRP).
- In addition, an IRB-approved data and safety monitoring
(DSM) plan must be sent (preferably electronically) to NHLBI
for
- All clinical
trials regardless of phase, and
- All clinical
research with greater than minimal
risk.
NOTE:
If an IRB-approved DSM plan is not received, NHLBI may request
certification that the proposed research is neither a clinical
trial nor a study that involves greater than minimal risk
to participants. Such certification must be countersigned
by the Authorized
Organizational Representative (AOR) and submitted
to the designated NHLBI Grants
Management Officer (GMO) or Contracting
Officer (CO) for review and acceptance prior to
commencement of human subjects research. Principal
investigators (PIs) with questions about the adequacy
or appropriateness of their DSM plans are encouraged to
consult with their Program
Official or Project Officer (PO) before submission
to the IRB.
- The DSM plan describes oversight and monitoring to ensure
the safety of participants and the integrity of the data.
DSM plans should address the following essential elements:
- Monitoring entity or who will monitor the study—e.g.,
PI, independent monitoring group, data and safety monitoring
board (DSMB) or observational study monitoring board
(OSMB). For example, for studies with low risk, monitoring
by the principal investigator and the IRB may suffice;
for higher risk studies, a DSMB may be required.
- Procedures for 1) monitoring study safety, 2) minimizing
research-associated risk, 3) protecting the confidentiality
of participant data, and 4) identifying, reviewing,
and reporting adverse
events and unanticipated
problems to the IRB, NHLBI, and FDA (if applicable).
For further information, see:
- For multi-site studies, procedures to ensure compliance
with the monitoring plan and reporting requirements
across study sites.
- An independent DSMB is generally required for phase
III clinical trials. A DSMB or OSMB may be required
for the following:
- Phase I or phase II
clinical trials that have multiple sites, are blinded,
or include high-risk interventions or vulnerable populations.
- Clinical research involving high-risk procedures/tests
or vulnerable populations.
NOTE: PIs are encouraged to consult with
their PO
for assistance in determining whether a DSMB/OSMB is required
for a given study. For further information, see NIH Policy
for Data and Safety Monitoring (http://grants.nih.gov/grants/guide/notice-files/not98-084.html)
and Further Guidance on Data and Safety Monitoring for Phase
I and Phase II Trials (http://grants.nih.gov/grants/guide/notice-files/NOT-OD-00-038.html).
- The PI must provide timely reporting
to the PO
of the following:
- Unanticipated problems or unexpected serious adverse
events that may be related to the study protocol.
- IRB-approved protocol or consent form revisions that
indicate a change in risk for participants.
- A summary of recommendations made by the DSMB/OSMB or
other monitoring entity and (if applicable) the action plan
for response.
- Notice of any actions taken by the IRB or regulatory
bodies regarding the research and any responses to those
actions.
NOTE: All personal identifiers must be
removed from any documents sent to NHLBI.
- In annual progress reports, PIs
should address the following:
- Adherence to the DSM plan.
- A summary of any DSM issues that have occurred during
the previous year.
- Any changes in the human subjects research or to the
DSM plan that affect risk. Modifications to the human subjects
research or DSM plan should be submitted to the NHLBI GMO
or CO prior to implementation of the change in study practice.
- New and continuing IRB approvals.
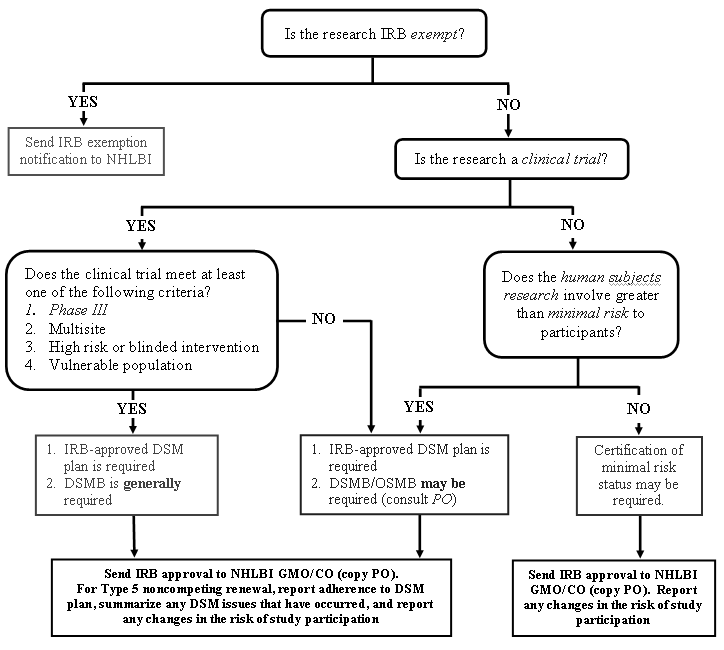
DSM Decision Flowchart for Research Involving Humans
or Human-Derived Materials
References:
Code of Federal Regulations, Title 45 - Public Welfare, Department
of Health and Human Services, Part 46 - Protection of Human
Subjects, 6/23/2005: http://www.hhs.gov/ohrp/humansubjects/guidance/45cfr46.htm.
Grants.gov Application Guide SF424 (R&R), Part II: Supplemental
Instructions for Preparing the Human Participants Section
of the Research Plan, 1/15/2008: http://grants1.nih.gov/grants/funding/424/index.htm.
NIH Policy for Data and Safety Monitoring, 6/10/1998: http://grants.nih.gov/grants/guide/notice-files/not98-084.html.
NIH Further Guidance on Data and Safety Monitoring for Phase
I and Phase II studies, 6/5/2000: http://grants.nih.gov/grants/guide/notice-files/NOT-OD-00-038.html.
NIH Guidance on Reporting Adverse Events to Institutional
Review Boards for NIH-Supported Multicenter Clinical Trials,
6/11/1999: http://grants1.nih.gov/grants/guide/notice-files/not99-107.html.
NHLBI Adverse Events and Unanticipated Problems Reporting
and Management (Interim Policy), May 2007: http://public.nhlbi.nih.gov/ocr/home/GetPolicy.aspx?id=16
OHRP Guidance on Reviewing and Reporting Unanticipated Problems
Involving Risks to Subjects or Others and Adverse Events,
1/15/2007: http://www.hhs.gov/ohrp/policy/AdvEvntGuid.htm.
Definitions:
Adverse
Event: As defined by OHRP,
an adverse event is any untoward or unfavorable medical occurrence
in a human subject, including any abnormal sign (for example,
abnormal physical exam or laboratory finding), symptom, or
disease, temporally associated with the subject’s participation
in the research, whether or not considered related to the
subject’s participation in the research. Adverse events
encompass both physical and psychological harms.
Authorized
Organizational Representative (AOR): The individual
authorized by the applicant organization to act for the applicant
and to assume the obligations imposed by the Federal laws,
regulations, requirements, and conditions that apply to grant
applications or grant awards. This official is equivalent
to the Signing Official (SO) in NIH’s eRA Commons. Responsibilities
include:
• Submitting the grant on behalf of the company, organization,
institution, or Government.
• Signing grant applications and the required certifications
and/or assurances necessary to fulfill the requirements of
the application process.
Award:
The provision of funds by NIH, based on an approved application
and budget or progress report, to an organizational entity
or an individual to carry out a project or activity.
Awardee: An institution receiving
a grant, cooperative agreement, or contract.
Clinical
Trial: A biomedical or behavioral research study
of human subjects designed to answer specific questions about
biomedical or behavioral interventions (drugs, treatments,
devices, or new ways of using known drugs, treatments, or
devices). Clinical trials are used to determine whether new
biomedical or behavioral interventions are safe, efficacious,
and effective. Clinical trials of an experimental drug, treatment,
device, or intervention may proceed through four phases:
Phase I. Testing in a small group of
people (e.g. 20-80) to determine efficacy and evaluate safety
(e.g., determine a safe dosage range and identify side effects).
Phase II. Study in a larger group of people (several hundred)
to determine efficacy and further evaluate safety.
Phase III. Study to determine efficacy in large groups of
people (from several hundred to several thousand) by comparing
the intervention to other standard or experimental interventions,
to monitor adverse effects, and to collect information to
allow safe use.
Phase IV. Studies done after the intervention has been marketed.
These studies are designed to monitor the effectiveness of
the approved intervention in the general population and to
collect information about any adverse effects associated with
widespread use.
Clinical
Research: Patient-oriented research, including epidemiologic
and behavioral studies, outcomes research, and health services
research. Patient-oriented research is research conducted
with human subjects (or on material of human origin such as
tissues, specimens, and cognitive phenomena) in which a researcher
directly interacts with human subjects. It includes research
on mechanisms of human disease, therapeutic interventions,
clinical trials, and development of new technologies, but
does not include in vitro studies using human tissues not
linked to a living individual. Studies falling under 45 CFR
46.101(a) (4) are not considered clinical research for purposes
of this definition.
Contract:
An award instrument establishing a binding legal agreement
between NIH and an award recipient (awardee) obligating the
latter to furnish products or services defined in detail by
NIH and binding the Institute to pay for them. Go to the Office
of Acquisition Management and Policy (OAMP) web site
for information on contracts and contract opportunities.
Contracting
Officer: Government employee authorized to execute
contractual agreements and to obligate funds on behalf of
the Government.
Cooperative
Agreement: Financial assistance mechanism used when
there will be substantial Federal scientific or programmatic
involvement. Substantial involvement means that, after award,
scientific or program staff will assist, guide, coordinate,
or participate in project activities.
Exempt
Research: As stated in 45 CFR 46.101(b): Unless otherwise
required by department or agency heads, research activities
in which the only involvement of human subjects will be in
one or more of the following categories are exempt from this
policy:
- Research conducted in established or commonly accepted
educational settings, involving normal educational practices,
such as (i) research on regular and special education instructional
strategies, or (ii) research on the effectiveness of or
the comparison among instructional techniques, curricula,
or classroom management methods.
- Research involving the use of educational tests (cognitive,
diagnostic, aptitude, achievement), survey procedures, interview
procedures or observation of public behavior, unless:
- (i) Information obtained is recorded in such a manner
that human subjects can be identified, directly or through
identifiers linked to the subjects; and (ii) any disclosure
of the human subjects' responses outside the research
could reasonably place the subjects at risk of criminal
or civil liability or be damaging to the subjects' financial
standing, employability, or reputation.
- Research involving the use of educational tests (cognitive,
diagnostic, aptitude, achievement), survey procedures, interview
procedures, or observation of public behavior that is not
exempt under paragraph (b)(2) of this section, if:
- ((i) The human subjects are elected or appointed public
officials or candidates for public office; or (ii) federal
statute(s) require(s) without exception that the confidentiality
of the personally identifiable information will be maintained
throughout the research and thereafter.
- Research, involving the collection or study of existing
data, documents, records, pathological specimens, or diagnostic
specimens, if these sources are publicly available or if
the information is recorded by the investigator in such
a manner that subjects cannot be identified, directly or
through identifiers linked to the subjects.
- Research and demonstration projects which are conducted
by or subject to the approval of department or agency heads,
and which are designed to study, evaluate, or otherwise
examine:
- ((i) Public benefit or service programs; (ii) procedures
for obtaining benefits or services under those programs;
(iii) possible changes in or alternatives to those programs
or procedures; or (iv) possible changes in methods or
levels of payment for benefits or services under those
programs.
- Taste and food quality evaluation and consumer acceptance
studies, (i) if wholesome foods without additives are consumed
or (ii) if a food is consumed that contains a food ingredient
at or below the level and for a use found to be safe, or
agricultural chemical or environmental contaminant at or
below the level found to be safe, by the Food and Drug Administration
or approved by the Environmental Protection Agency or the
Food Safety and Inspection Service of the U.S. Department
of Agriculture.
Grant:
Financial assistance mechanism providing money, property,
or both to an eligible entity to carry out an approved project
or activity. A grant is used whenever the NIH Institute or
Center anticipates no substantial programmatic involvement
with the recipient during performance of the financially assisted
activities.
Grantee:
The organization or individual awarded a grant or cooperative
agreement by NIH that is responsible and accountable for the
use of the funds provided and for the performance of the grant-supported
project or activities. The grantee is the entire legal entity
even if a particular component is designated in the award
document. The grantee is legally responsible and accountable
to NIH for the performance and financial aspects of the grant-supported
project or activity.
Grants
Management Officer (GMO): An NIH official responsible
for the business management aspects of grants and cooperative
agreements, including review, negotiation, award, and administration,
and for the interpretation of grants administration policies
and provisions. Only GMOs are authorized to obligate NIH to
the expenditure of funds and permit changes to approved projects
on behalf of NIH. Each NIH Institute and Center awarding grants
has one or more GMOs with responsibility for particular programs
or awards.
Human
Subject: A living individual about whom an investigator
(whether professional or student) conducting research obtains
data through intervention or interaction with the individual
or obtains identifiable private information. Regulations governing
the use of human subjects in research extend to use of human
organs, tissues, and body fluids from identifiable individuals
as human subjects and to graphic, written, or recorded information
derived from such individuals.
Human
Subjects Research: A systematic investigation that
includes human subjects.
Minimal Risk: As defined in
45
CFR 46.102, minimal risk means that the probability
and magnitude of harm or discomfort anticipated in the research
are not greater in and of themselves than those ordinarily
encountered by the research population in daily life or during
the performance of routine physical or psychological examinations
or tests.
For example, minimal risk in a healthy population may include
venipuncture (blood draws), physical examinations, routine
psychological testing, nutritional studies, behavioral research,
survey/questionnaire studies, and observational research.
This category includes specimen-based studies that do not
qualify for an exemption and are considered to be clinical
research by the applicable IRB.
Notice
of Award (NoA): The legally binding document
• notifying the grantee and others that an award has
been made;
• containing or referencing all terms and conditions
of the award;
• documenting the obligation of Federal funds (may be
in letter format and may be issued electronically).
Previously known as Notice of Grant Award (NGA).
Office for Human Research
Protections (OHRP): HHS office overseeing human subject
protection for HHS-supported research. Go to OHRP.
Phase
III Clinical Trial: As defined by NIH, a broadly
based prospective investigation (usually involving several
hundred or more human subjects) to evaluate an experimental
intervention in comparison with a standard or control intervention
or to compare two or more existing treatments. The definition
includes pharmacologic, non-pharmacologic, and behavioral
interventions given for disease prevention, prophylaxis, diagnosis,
or therapy. Community trials and other population-based intervention
trials also are included.
Phases
of Clinical Trials: See Clinical
Trial
Program
Official: The NIH staff member responsible for the
programmatic, scientific, and/or technical aspects of a grant.
Note that this title is most frequently used to refer to the
scientific or technical representative for grants, but the
generic “program official” term also may refer
to a Project Officer if the funding mechanism is a contract.
Project
Officer: The NIH staff member designated as a Contracting
Officer’s technical representative to coordinate the
substantive aspects of an acquisition from its development
through to contract award and administration.
Unanticipated
Problem: According to OHRP, unanticipated problems
generally include any incident, experience, or outcome that
meets all of the following criteria:
(1) unexpected (in terms of nature, severity, or frequency)
given (a) the research procedures that are described in the
protocol-related documents, such as the IRB-approved research
protocol and informed consent document; and (b) the characteristics
of the subject population being studied;
(2) related or possibly related to participation in the research
(in this guidance document, possibly related means there is
a reasonable possibility that the incident, experience, or
outcome may have been caused by the procedures involved in
the research); and
(3) suggests that the research places subjects or others at
a greater risk of harm (including physical, psychological,
economic, or social harm) than was previously known or recognized.
FAQs :
1) Q. How does the NHLBI policy differ from the NIH policy?
A. The NHLBI policy is aligned with the NIH policy and extends
the requirement of DSM plans to include clinical research
with greater than minimal risk in addition to clinical trials.
The NHLBI policy explicitly requires IRB approval of the DSM
plan.
2) Q. Who determines whether my research protocol has
greater than minimal risk?
A. Your IRB has primary responsibility for determination
of the level of risk to study participants. When a decision
is made to fund your grant or contract, the PO will issue
instructions to release funds for your research protocol if
the study is clearly minimal risk, as defined in 45 CFR 46.102
(i). ( http://www.hhs.gov/ohrp/humansubjects/guidance/45cfr46.htm#46.102
.) If the PO considers your protocol to be of greater
than minimal risk, an IRB-approved DSM plan will be requested.
If you disagree with the PO 's assessment, you may be asked
for clarification or certification of your IRB's determination
of risk.
3) Q. Does this policy apply to all award mechanisms
used by NHLBI?
A. Yes, this policy applies to all NHLBI-sponsored research
projects regardless of whether they are contracts
, grants
, or cooperative
agreements .
4) Q. My data and safety monitoring plan is part of
my grant application. Is this sufficient, or am I required
to send something else?
A. NHLBI Policy requires an IRB-approved DSM plan. The DSM
plan in your grant application is sufficient if (1) it contains
the essential elements listed in the NHLBI policy and (2)
you can document that your IRB has reviewed and approved the
relevant section of your grant application.
5) Q. What happens if I don't provide a DSM plan in
my grant application?
A. If your application is selected for funding and is either
a clinical trial or clinical research entailing more than
minimal risk, NHLBI will place a term and condition on the
Notice
of Award (NoA) restricting all human subjects research
activities until the grantee
can demonstrate that the IRB has reviewed and approved
the DSM plan. A similar process is followed for contracts.
6) Q What if the human subjects aspect
of my research begins after the first year of funding?
A. This policy only applies to human
subjects research . For example, if the human subjects
research portion of your study begins in year 3, then this
policy must be followed prior to the commencement of human
subjects research activities starting in year 3. Other research
activities that do not involve human subjects may proceed.
See the terms and conditions of the award for further information.
7) Q. I am concerned about protecting participants'
privacy, as well as sending sensitive study data to the NHLBI
PO .
A. It is important that all personal identifiers be removed
from any documents before sending them to the NHLBI. In addition,
the reporting requirements specify that summary recommendations
should be sent to the PO . Please d o not send the
confidential or sensitive data upon which such recommendations
are based u nless specifically requested by the PO .
8) Q. Where and when do I send the data and safety monitoring
(DSM) plan or minimal risk certification?
A. If the project is a grant or cooperative agreement, all
official documentation must be sent (preferably electronically)
to the Grants
Management Officer —with a copy to the Program
Official —listed in eRA Commons ( https://commons.era.nih.gov/commons/
). If the project is a contract, this documentation should
be sent to the Contracting
Officer —with a copy to the Project
Officer —per the contract. Please send the documentation
anytime after the IRB has approved the DSM plan or indicated
that the research is minimal risk but BEFORE any human subjects
research commences. NHLBI also should be notified if the human
subjects research, DSM plan, or minimal risk designation changes
during the project period. This documentation must be countersigned
by the Authorized
Organizational Representative .
9) Q. What if I cannot submit the documentation electronically?
A. The documentation can be mailed or faxed to the applicable
Grants
Management Officer or Contracting
Officer (see question 7 above). The following Grants
Management central fax number will convert faxed documents
to PDF files: ( 301) 451-5462.
|