|
|
|
James Inglese, Ph.D.
|
|
Dr. Inglese's research interest is focused on the discovery and development of small molecule "biomodulators," compounds capable of affecting biological function in a defined in vitro setting. Small molecule biomodulators allow altering biological systems or target protein activities in a temporally controlled and dose-dependent manner, and in contrast to nucleic acid based tools such as siRNAs, they generally interact with proteins, the ultimate effectors of biological response.
Dr. Inglese arrived at NHGRI in May 2004 from Merck, and is currently overseeing infrastructure development of the National Institutes of Health (NIH) Chemical Genomics Center (NCGC). The NCGC is the first component of the NIH Roadmap Molecular Libraries Screening Center Network, a nationwide group of screening centers that will produce innovative biomodulators for use in the study of gene, cell and organismal function. Assays submitted to the NCGC will be screened against a diverse collection of >500,000 chemical compounds, including substances not previously tested because they were not "drug-like," such as natural products, cellular metabolites and biosynthetic intermediates. The identification and optimization of small molecule biomodulators requires the collection of data from thousands of individual assays, and involves a fusion of biology, automation, complex data analysis and chemistry. Thus, a multidisciplinary team of scientists, engineers and informatics experts is being assembled and, when fully developed, the NCGC will have approximately 50 scientists and support staff who will work together in an integrated biomolecular screening and profiling, chemistry and informatics research effort.
A veteran of the pharmaceutical and biotechnology industry, Dr. Inglese has developed a large number of assay methods and reagents, including one of the first high-sensitivity fluorescence G protein-coupled receptor assays, and chemical methodologies to facilitate labeling of ligands for HT radiometric assays. He pioneered the use of laser-scanning imaging, a technology that permits high-throughput cell or particle-based assays. Most recently, he explored the use of naturally occurring protein domains, in combination with protein evolution techniques, to create antibody surrogates for the detection of post-translationally modified peptides and proteins. Such engineered domains have been used successfully in the development of protease and phosphatase assays for high throughput screening (HTS).
|
Recent Publications:
|
Molecular Cancer Therapeutics
|
Identification of phosphotyrosine mimetic inhibitors of Human Tyrosyl-DNA Phosphodiesterase I by a novel AlphaScreen high-throughput assay.
Marchand C, Lea W, Jadhav A, Dexheimer T, Austin CP, Inglese J, Pommier Y, Simeonov A.
Tyrosyl-DNA phosphodiesterase I (Tdp1) resolves topoisomerase I (Top1)-DNA
adducts accumulated from natural DNA damage, as well as from the action of certain
anticancer drugs. Tdp1 catalyzes the hydrolysis of the phosphodiester bond between the
catalytic tyrosine residue of Top1 and the DNA 3’ phosphate. Only a limited number of
weak inhibitors have been reported for Tdp1, and there is an unmet need to identify novel
chemotypes through screening of chemical libraries. Herein we present an easily
configured, highly-miniaturized, and robust Tdp1 assay utilizing the AlphaScreen
technology. Uninhibited enzyme reaction is associated with low signal while inhibition
leads to a gain of signal, making the present assay format especially attractive for
automated large-collection high-throughput screening. We report the identification and
initial characterization of four previously-unreported inhibitors of Tdp1. Among them,
suramin, NF449 and methyl-3,4-dephostatin are phosphotyrosine mimetics that may act
as Tdp1 substrate decoys. We also report a novel biochemical assay using the SCAN1
Tdp1 mutant to study the mechanism of action of methyl-3,4 dephostatin.
|
|
Combinatorial Chemistry & High Throughput Screening
|
Optimization and Validation of Two Miniaturized Glucocerebrosidase Enzyme Assays for High Throughput Screening
Urban DJ, Zheng W, Goker-Alpan O, Jadhav A, LaMarca ME, Inglese J, Sidransky E, Austin CP
Glucocerebrosidase (GC) catalyzes the hydrolysis of ß-glucocerebroside to glucose and ceramide in lysosomes. Mutations in the glucocerebrosidase gene (GBA) result in Gaucher disease, an autosomal recessive lysosomal storage disorder. Many of the mutations encountered in patients with Gaucher disease are missense alterations that may cause misfolding, decreased stability and/or mistrafficking of this lysosomal protein. Some inhibitors of GC have been shown to act as chemical chaperones, stabilizing the conformation of mutant proteins and thus restoring their function. High throughput screening (HTS) of small molecule libraries for such compounds with potential for chaperone therapy requires an accurate, reproducible and sensitive assay method. We have adapted and optimized two fluorogenic GC enzyme assays and miniaturized them into the 1536-well plate format for HTS. The two substrates, 4-methylumbelliferyl ß-D-glucopyranoside and resorufin ß-D-glucopyranoside, have Km values of 768 µM and 33 µM, respectively, and different emission spectra. Paired screening with the two assays helps to eliminate false inference of activity due to autofluorescence or fluorescence quenching by the screened compounds. Test screens with the LOPAC library indicated that both assays were robust for HTS, and gave comparable results for GC inhibitor activities. These two assays can be used to identify both GC activators and inhibitors with potential therapeutic value.
|
ASSAY and Drug Development Technologies

|
A robotic platform for quantitative high-throughput screening.
Michael S, Auld D, Klumpp C, Jadhav A, Zheng W, Thorne N, Austin CP, Inglese J, Simeonov A.
High-throughput screening (HTS) is increasingly being adopted in academic institutions, where the decoupling of screening and drug development has led to unique challenges, as well as novel uses of instrumentation, assay formulations, and software tools. Advances in technology have made automated unattended screening in the 1,536-well plate format broadly accessible and have further facilitated the exploration of new technologies and approaches to screening. A case in point is our recently developed quantitative HTS (qHTS) paradigm, which tests each library compound at multiple concentrations to construct concentration-response curves (CRCs) generating a comprehensive data set for each assay. The practical implementation of qHTS for cell-based and biochemical assays across libraries of > 100,000 compounds (e.g., between 700,000 and 2,000,000 sample wells tested) requires maximal efficiency and miniaturization and the ability to easily accommodate many different assay formats and screening protocols. Here, we describe the design and utilization of a fully integrated and automated screening system for qHTS at the National Institutes of Health's Chemical Genomics Center. We report system productivity, reliability, and flexibility, as well as modifications made to increase throughput, add additional capabilities, and address limitations. The combination of this system and qHTS has led to the generation of over 6 million CRCs from > 120 assays in the last 3 years and is a technology that can be widely implemented to increase efficiency of screening and lead generation.
|
ACS Chemical Biology

|
A Specific Mechanism for Nonspecific Activation in Reporter-Gene Assays.
Auld DS, Thorne N, Nguyen DT, Inglese J.
The importance of bioluminescence in enabling a broad range of high-throughput screening (HTS) assay formats is evidenced by widespread use in industry and academia. Therefore, understanding the mechanisms by which reporter enzyme activity can be modulated by small molecules is critical to the interpretation of HTS data. In this Perspective, we provide evidence for stabilization of luciferase by inhibitors in cell-based luciferase reporter-gene assays resulting in the counterintuitive phenomenon of signal activation. These data were derived from our analysis of luciferase inhibitor compound structures and their prevalence in the Molecular Libraries Small Molecule Repository using 100 HTS experiments available in PubChem. Accordingly, we found an enrichment of luciferase inhibitors in luciferase reporter-gene activation assays but not in assays using other reporters. In addition, for several luciferase inhibitor chemotypes, we measured reporter stabilization and signal activation in cells that paralleled the inhibition determined using purified luciferase to provide further experimental support for these contrasting effects.
|
|
Journal of Medicinal Chemistry
|
Characterization of Chemical Libraries for Luciferase Inhibitory Activity.
Auld DS, Southall N, Jadhav A, Johnson RL, Diller D, Simeonov S, Austin CP, Inglese J.
To aid in the interpretation of HTS results derived from luciferase-based assays we used quantitative HTS (qHTS), an approach that defines the concentration-response behavior of each library sample, to profile the ATP-dependent luciferase from Photinus pyralis against >70,000 samples. We found approximately 3% of the library was active, containing only compounds with inhibitory concentration-responses of which 681 (0.9%) exhibited IC50s < 10 uM. Representative compounds were shown to inhibit purified P. pyralis as well as several commercial luciferase-based detection reagents but were found to be largely inactive against Renilla reniformis luciferase. Light attenuation by the samples was also examined and found to be more prominent in the blue-shifted bioluminescence produced by R. reniformas luciferase than with bioluminescence produced by P. pyralis luciferase. We describe the SAR of the luciferase inhibitors and discuss the use of this data in the interpretation of HTS results, and configuration of luciferase-based assays.
|
|
Journal of Medicinal Chemistry
|
Fluorescence Spectroscopic Profiling of Compound Libraries.
Simeonov S, Jadhav A, Thomas CJ, Wang Y, Huang R, Southall N, Shinn P, Smith J, Austin CP, Inglese J.
Chromo/fluorophoric properties often accompany the conjugated, aromatic and heterocyclic features of many of the scaffolds and impurities that make up library samples used for high throughput screening (HTS). These properties impart highly variable effects on assay outputs employing optical detection, thus complicating the interpretation of data and leading to false positives and negatives. Here, we report the comprehensive fluorescence profile of >70,000 samples across multiple spectral regions commonly utilized in HTS assays. The quantitative HTS (qHTS) paradigm was utilized to test each sample at seven or more concentration points over a 4-log concentration range in 1536-well format, with raw fluorescence response collected using a CCD-based imager. The resulting output was compared with fluorophore standards to compute a normalized fluorescence response (termed fluorophore-equivalent concentration, FEC) for each sample, concentration, and relevant spectral region. The greatest fraction of fluorescent compounds appeared in the UV-end of the light spectrum, where over 5% of library members matched or exceeded 10 nM FEC of 4-methylumbelliferone and AlexaFluor 350, while approximately 1.8% of the library matched or exceeded 100 nM FEC of these standards. Red-shifting the spectral window by as little as 100 nm was accompanied by a dramatic decrease in autofluorescence. Native compound fluorescence, scaffold overlap with known fluorophores, fluorescent impurities, novel fluorescent compounds, and the ability to discriminate generalities of fluorescent interferences and devise strategies to identify them are discussed.
|
|
Combinatorial Chemistry & High Throughput Screening
|
A miniaturized glucocorticoid receptor translocation assay using enzymatic fragment complementation evaluated with qHTS.
Zhu PJ, Zheng W, Auld DS, Jadhav A, Macarthur R, Olson KR, Peng K, Dotimas H, Austin CP, Inglese J.
Nuclear translocation is an important step in glucocorticoid receptor (GR) signaling and assays that measure this process allow the identification of nuclear receptor ligands independent of subsequent functional effects. To facilitate the identification of GR-translocation agonists, an enzyme fragment complementation (EFC) cell-based assay was scaled to a 1536-well plate format to evaluate 9,920 compounds using a quantitative high throughput screening (qHTS) strategy where compounds are assayed at multiple concentrations. In contrast to conventional assays of nuclear translocation the qHTS assay described here was enabled on a standard luminescence microplate reader precluding the requirement for imaging methods. The assay uses beta-galactosidase alpha complementation to indirectly detect GR-translocation in CHO-K1 cells. 1536-well assay miniaturization included the elimination of a media aspiration step, and the optimized assay displayed a Z' of 0.55. qHTS yielded EC(50) values for all 9,920 compounds and allowed us to retrospectively examine the dataset as a single concentration-based screen to estimate the number of false positives and negatives at typical activity thresholds. For example, at a 9 microM screening concentration, the assay showed an accuracy that is comparable to typical cell-based assays as judged by the occurrence of false positives that we determined to be 1.3% or 0.3%, for a 3sigma or 6sigma threshold, respectively. This corresponds to a confirmation rate of approximately 30% or approximately 50%, respectively. The assay was consistent with glucocorticoid pharmacology as scaffolds with close similarity to dexamethasone were identified as active, while, for example, steroids that act as ligands to other nuclear receptors such as the estrogen receptor were found to be inactive.
|
|
ASSAY and Drug Development Technologies
|
A 1,536-Well-Based Kinetic HTS Assay for Inhibitors of Schistosoma mansoni Thioredoxin Glutathione Reductase.
Lea WA, Jadhav A, Rai G, Sayed AA, Cass CL, Inglese J, Williams DL, Austin CP, Simeonov A.
Schistosomiasis is a major neglected tropical disease that currently affects over 200 million people and leads to over 200,000 annual deaths. Schistosoma mansoni parasites survive in humans in part because of a set of antioxidant enzymes that continuously degrade reactive oxygen species produced by the host. A principal component of this defense system has been recently identified as thioredoxin glutathione reductase (TGR), a parasite-specific enzyme that combines the functions of two human counterparts, glutathione reductase and thioredoxin reductase, and as such this enzyme presents an attractive new target for anti-schistosomiasis drug development. Herein, we present the development of a highly miniaturized and robust screening assay for TGR. The 5-mul final volume assay is based on the Ellman reagent [5,5'-dithiobis(2-nitrobenzoic acid) (DTNB)] and utilizes a high-speed absorbance kinetic read to minimize the effect of dust, absorbance interference, and meniscus variation. This assay is further applicable to the testing of other redox enzymes that utilize DTNB as a model substrate.
|
|
Journal of Biomolecular Screening
|
A Cell-Based PDE4 Assay in 1536-Well Plate Format for High-Throughput Screening.
Titus SA, Xiao L, Southall N, Lu J, Inglese J, Brasch M, Austin CP, Zheng W.
The cyclic nucleotide phosphodiesterases (PDEs) are intracellular enzymes that catalyze the hydrolysis of 3,'5'-cyclic nucleotides, such as cyclic adenosine monophosphate (cAMP) and cyclic guanosine monophosphate (cGMP), to their corresponding 5'nucleotide monophosphates. These enzymes play an important role in controlling cellular concentrations of cyclic nucleotides and thus regulate a variety of cellular signaling events. PDEs are emerging as drug targets for several diseases, including asthma, cardiovascular disease, attention-deficit hyperactivity disorder, Parkinson's disease, and Alzheimer's disease. Although biochemical assays with purified recombinant PDE enzymes and cAMP or cGMP substrate are commonly used for compound screening, cell-based assays would provide a better assessment of compound activity in a more physiological context. The authors report the development and validation of a new cell-based PDE4 assay using a constitutively active G-protein-coupled receptor as a driving force for cAMP production and a cyclic nucleotide-gated cation channel as a biosensor in 1536-well plates.
|
Nature Medicine

|
Identification of oxadiazoles as new drug leads for the control of schistosomiasis
Sayed AA, Simeonov A, Thomas CJ, Inglese J, Austin CP, Williams DL.
Schistosomiasis is a tropical disease associated with high morbidity and mortality, currently affecting over 200 million people worldwide. Praziquantel is the only drug used to treat the disease, and with its increased use the probability of developing drug resistance has grown significantly. The Schistosoma parasites can survive for up to decades in the human host due in part to a unique set of antioxidant enzymes that continuously degrade the reactive oxygen species produced by the host's innate immune response. Two principal components of this defense system have been recently identified in S. mansoni as thioredoxin/glutathione reductase (TGR) and peroxiredoxin (Prx) and as such these enzymes present attractive new targets for anti-schistosomiasis drug development. Inhibition of TGR/Prx activity was screened in a dual-enzyme format with reducing equivalents being transferred from NADPH to glutathione via a TGR-catalyzed reaction and then to hydrogen peroxide via a Prx-catalyzed step. A fully automated quantitative high-throughput (qHTS) experiment was performed against a collection of 71,028 compounds tested as 7- to 15-point concentration series at 5 microL reaction volume in 1536-well plate format. In order to generate a robust data set and to minimize the effect of compound autofluorescence, apparent reaction rates derived from a kinetic read were utilized instead of end-point measurements. Actives identified from the screen, along with previously untested analogues, were subjected to confirmatory experiments using the screening assay and subsequently against the individual targets in secondary assays. Several novel active series were identified which inhibited TGR at a range of potencies, with IC(50)s ranging from micromolar to the assay response limit ( approximately 25 nM). This is, to our knowledge, the first report of a large-scale HTS to identify lead compounds for a helminthic disease, and provides a paradigm that can be used to jump-start development of novel therapeutics for other neglected tropical diseases.
|
|
Toxicology in Vitro
|
A bioluminescent cytotoxicity assay for assessment of membrane integrity using a proteolytic biomarker.
Cho MH, Niles A, Huang R, Inglese J, Austin CP, Riss T, Xia M.
Measurement of cell membrane integrity has been widely used to assess chemical cytotoxity. Several assays are available for determining cell membrane integrity including differential labeling techniques using neutral red and trypan blue dyes or fluorescent compounds such as propidium iodide. Other common methods for assessing cytotoxicity are enzymatic "release" assays which measure the extra-cellular activities of lactate dehydrogenase (LDH), adenylate kinase (AK), or glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in culture medium. However, all these assays suffer from several practical limitations, including multiple reagent additions, scalability, low sensitivity, poor linearity, or requisite washes and medium exchanges. We have developed a new cytotoxicity assay which measures the activity of released intracellular proteases as a result of cell membrane impairment. It allows for a homogenous, one-step addition assay with a luminescent readout. We have optimized and miniaturized this assay into a 1536-well format, and validated it by screening a library of known compounds from the National Toxicology Program (NTP) using HEK 293 and human renal mesangial cells by quantitative high-throughput screening (qHTS). Several known and novel membrane disrupters were identified from the library, which indicates that the assay is robust and suitable for large scale library screening. This cytotoxicity assay, combined with the qHTS platform, allowed us to quickly and efficiently evaluate compound toxicities related to cell membrane integrity.
|
Proceedings of the National Academy of Sciences

|
Three classes of glucocerebrosidase inhibitors identified by quantitative high-throughput screening are chaperone leads for Gaucher disease
Zheng W, Padia J, Urban D, Jadhav A, Simeonov A, Goldin E, Auld DS, LaMarca ME, Inglese J, Austin CP, Sidransky E.
Gaucher disease is an autosomal recessive lysosomal storage disorder caused by mutations in the glucocerebrosidase gene. Missense mutations result in reduced enzyme activity that may be due to misfolding, raising the possibility of small-molecule chaperone correction of the defect. Screening large compound libraries by quantitative high-throughput screening (qHTS) provides comprehensive information on the potency, efficacy, and structure-activity relationships (SAR) of active compounds directly from the primary screen, facilitating identification of leads for medicinal chemistry optimization. We used qHTS to rapidly identify three structural series of potent, selective, nonsugar glucocerebrosidase inhibitors. The three structural classes had excellent potencies and efficacies and, importantly, high selectivity against closely related hydrolases. Preliminary SAR data were used to select compounds with high activity in both enzyme and cell-based assays. Compounds from two of these structural series increased N370S mutant glucocerebrosidase activity by 40-90% in patient cell lines and enhanced lysosomal colocalization, indicating chaperone activity. These small molecules have potential as leads for chaperone therapy for Gaucher disease, and this paradigm promises to accelerate the development of leads for other rare genetic disorders.
|
Current Chemical Genomics
|
Comparison on Functional Assays for Gq-Coupled GPCRs by Measuring Inositol Monophospate-1 and Intracellular Calcium in 1536-Well Plate Format
Liu K, Titus SA, Southall N, Zhu P, Inglese J, Austin CP, Zheng w.
Cell-based functional assays used for compound screening and lead optimization play an important role in drug discovery for G-protein coupled receptors (GPCRs). Cell-based assays can define the role of a compound as an agonist, antagonist or inverse agonist and can provide detailed information about the potency and efficacy of a compound. In addition, cell-based screens can be used to identify allosteric modulators that interact with sites other than the binding site of the endogenous ligand. Intracellular calcium assays which use a fluorescent calcium binding dye (such as Fluo-3, Fluo-4 or Fura-2) have been used in compound screening campaigns to measure the activity of Gq-coupled GPCRs. However, such screening methodologies require a special instrumentation to record the rapid change in intracellular free calcium concentration over time. The radioactive inositol 1,4,5- triphosphate (IP3) assay measures 3H-inositol incorporation and is another traditional assay for the assessment of Gq-coupled GPCR activity, but it is not suitable for screening of large size compound collections because it requires a cell wash step and generates radioactive waste. To avoid these limitations, we have optimized and miniaturized a TR-FRET based IP-One assay that measures inositol monophosphate in a 1536-well plate format. This assay is homogenous, non-radioactive and does not require a kinetic readout. It has been tested with the cell lines expressing M1 acetylcholine, FFAR1, vasopressin V1b, or Neuropeptide S receptors. The activities of antagonists determined in the IP-One assay correlated well with these measured in the intracellular calcium assay while the correlation of agonist activities might vary from cell line to cell line. This IP-One assay offers an alternative method for high throughput screening of Gq-coupled GPCRs without using costly kinetic plate readers.
|
|
PLoS Neglected Tropical Diseases
|
Quantitative High-Throughput Screen Identifies Inhibitors of the Schistosoma mansoni Redox Cascade.
Simeonov S, Jadhav A, Sayed AA, Wang Y, Nelson ME, Inglese J, Williams DL, Austin CP.
Schistosomiasis is a tropical disease associated with high morbidity and mortality, currently affecting over 200 million people worldwide. Praziquantel is the only drug used to treat the disease and with its increased use the probability of developing drug resistance has grown significantly. The Schistosoma parasites can survive for up to decades in the human host due in part to a unique set of antioxidant enzymes that continuously degrade the reactive oxygen species produced by the host's innate immune response. Two principle components of this defense system have been recently identified in S. mansoni as thioredoxin/glutathione reductase (TGR) and peroxiredoxin (Prx) and as such these enzymes present attractive new targets for anti-schistosomiasis drug development. Inhibition of TGR/Prx activity was screened in a dual-enzyme format with reducing equivalents being transferred from NADPH to a glutathione intermediate via a TGR-catalyzed reaction and then to hydrogen peroxide via Prx-catalyzed step. A fully-automated qHTS experiment (Inglese et al, PNAS, 103, 1147 (2006)) was performed against a collection of 71,028 compounds tested as 7- to 15-point concentration series at 5 ?L reaction volume in 1536-well plate format. In order to generate a robust data set and to minimize the effect of compound autofluorescence, apparent reaction rates derived from a kinetic read were utilized instead of end-point measurements. Actives identified from the screen, along with previously-untested analogues, were subjected to confirmatory experiments using the screening assay and subsequently against the individual targets in secondary assays. Several novel active series were identified which inhibited TGR at a range of potencies, with IC50s ranging from micromolar to the assay response limit (~25 nM). This is, to our knowledge, the first report of a large-scale HTS to identify lead compounds for a helminthic disease.
|
|
Bioorganic & Medicinal Chemistry Letters
|
Identification of a potent new chemotype for the selective inhibition of PDE4.
Skoumbourdis AP, Huang R, Southall N, Leister W, Guo V, Cho MH, Inglese J, Nirenberg M, Austin CP, Xia M, Thomas CJ.
A series of substituted 3,6-diphenyl-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazines were prepared and analyzed as inhibitors of phosphodiesterase 4 (PDE4). Synthesis, structure–activity relationships, and the selectivity of a highly potent analogue against related phosphodiesterase isoforms are presented.

|
Chemical Research Toxicology

|
Characterization of Diversity in Toxicity Mechanism Using In Vitro Cytotoxicity Assays in Quantitative High Throughput Screening.
Huang R, Southall N, Cho MH, Xia M, Inglese J, Austin CP.
Assessing the potential health risks of environmental chemical compounds is an expensive
undertaking which has motivated the development of new alternatives to traditional in vivo
toxicological testing. One approach is to stage the evaluation, beginning with less expensive and
higher throughput in vitro testing before progressing to more definitive trials. In vitro testing can
be used to generate a hypothesis about a compound's mechanism of action, which can then be
used to design an appropriate in vivo experiment. Here we begin to address the question of how
to design such a battery of in vitro cell-based assays by combining data from two different types
of assays, cell viability and caspase activation, with the aim of elucidating mechanism of action.
Because caspase activation is a transient event during apoptosis, it is not possible to design a
single end-point assay protocol that would identify all instances of compound-induced caspase
activation. Nevertheless, useful information about compound mechanism of action can be
obtained from these assays in combination with cell viability data. Unsupervised clustering in
combination with Dunn's cluster validity index is a robust method for identifying mechanisms of
action without requiring any a priori knowledge about mechanisms of toxicity. The performance
of this clustering method is evaluated by comparing the clustering results against literature
annotations of compound mechanisms.
|
|
Combinatorial Chemistry & High Throughput Screening
|
A High Throughput Fluorescence Polarization Assay for Inhibitors of the GoLoco Motif/G-alpha Interaction.
Kimple AJ, Yasgar A, Hughes M, Jadhav A, Willard FS, Muller RE, Austin CP, Inglese J, Ibeanu GC, Siderovski DP, Simeonov A.
The GoLoco motif is a short Galpha-binding polypeptide sequence. It is often found in proteins that regulate cell-surface receptor signaling, such as RGS12, as well as in proteins that regulate mitotic spindle orientation and force generation during cell division, such as GPSM2/LGN. Here, we describe a high throughput fluorescence polarization (FP) assay using fluorophore-labeled GoLoco motif peptides for identifying inhibitors of the GoLoco motif interaction with the G-protein alpha subunit Galpha (i1). The assay exhibits considerable stability over time and is tolerant to DMSO up to 5%. The Z'-factors for robustness of the GPSM2 and RGS12 GoLoco motif assays in a 96-well plate format were determined to be 0.81 and 0.84, respectively; the latter assay was run in a 384-well plate format and produced a Z'-factor of 0.80. To determine the screening factor window (Z-factor) of the RGS12 GoLoco motif screen using a small molecule library, the NCI Diversity Set was screened. The Z-factor was determined to be 0.66, suggesting that this FP assay would perform well when developed for 1,536-well format and scaled up to larger libraries. We then miniaturized to a 4 microL final volume a pair of FP assays utilizing fluorescein- (green) and rhodamine- (red) labeled RGS12 GoLoco motif peptides. In a fully-automated run, the Sigma-Aldrich LOPAC(1280) collection was screened three times with every library compound being tested over a range of concentrations following the quantitative high throughput screening (qHTS) paradigm; excellent assay performance was noted with average Z-factors of 0.84 and 0.66 for the green- and red-label assays, respectively.
|
|
Analytical Biochemistry
|
Dual-fluorophore quantitative high-throughput screen for inhibitors of BRCT-phosphoprotein interaction.
Simeonov A, Yasgar A, Jadhav A, Lokesh GL, Klumpp C, Michael S, Inglese J, Austin CP, Natarajan A.
Finding specific small-molecule inhibitors of protein-protein interactions remains a significant challenge. Recently, attention has grown toward "hot spot" interactions where binding is dominated by a limited number of amino acid contacts, theoretically offering an increased opportunity for disruption by small molecules. Inhibitors of the interaction between BRCT (the C-terminal portion of BRCA1, a key tumor suppressor protein with various functions) and phosphorylated proteins (Abraxas/BACH1/CtIP), implicated in DNA damage response and repair pathways, should prove to be useful in studying BRCA1's role in cancer and in potentially sensitizing tumors to chemotherapeutic agents. We developed and miniaturized to a 1536-well format and 3ul final volume a pair of fluorescence polarization (FP) assays using fluorescein- and rhodamine-labeled pBACH1 fragment. To minimize the effect of fluorescence artifacts and to increase the overall robustness of the screen, the 75,552 compound library members all were assayed against both the fluorescein- and rhodamine-labeled probe-protein complexes in separate but interleaved reactions. In addition, every library compound was tested over a range of concentrations following the quantitative high-throughput screening (qHTS) paradigm. Analyses of the screening results led to the selection and subsequent confirmation of 16 compounds active in both assays. Faced with a traditionally difficult protein-protein interaction assay, by performing two-fluorophore qHTS, we were able to confidently select a number of actives for further studies.
|
|
Analytical Biochemistry
|
A Quantitative High-Throughput Screen Identifies Potential Epigenetic Modulators of Gene Expression.
Johnson RL, Huang W, Jadhav A, Austin CP, Inglese J, Martinez ED.
Epigenetic regulation of gene expression is essential in embryonic development and
contributes to cancer pathology. We used a cell-based imaging assay that measures
derepression of a silenced GFP reporter to identify novel classes of compounds
involved in epigenetic regulation. This Locus Derepression (LDR) assay was screened
against a 69,137-member chemical library using quantitative high-throughput screening
(qHTS), a titration-response method that assays compounds at multiple concentrations.
From structure-activity relationships of the 411 actives recovered from the qHTS, six
distinct chemical series were chosen for further study. Forty-eight qHTS actives and
analogs were counter screened using the parental line of the LDR cells, which lack the
GFP reporter. Three series, 8-hydroxy quinoline, quinoline-8-thiol and 1,3,5-
thiadiazinane-2-thione, were not fluorescent and re-confirmed activity in the LDR cells.
The three active series did not inhibit histone deacetylase activity in nuclear extracts or
reactivate the expression of the densely methylated p16 gene in cancer cells. However,
one series induced expression of the methylated CDH13 gene and inhibited the viability
of several lung cancer lines at submicromolar concentrations. These results suggest
that the identified small molecules act on epigenetic or transcriptional components and
validate our approach of using a cell-based imaging assay in conjunction with qHTS.
|
|
Journal of Medicinal Chemistry
|
A Comprehensive Mechanistic Analysis of Hits from High-Throughput and Docking Screens Against Beta-Lactamase.
Babaoglu K, Simeonov A, Irwin, J, Nelson M, Feng BY, Thomas C, Cancian L, Costi MP, Maltby D, Jadhav A, Inglese J, Austin CP, Shoichet BK.
High-throughput screening (HTS) is widely used in drug discovery. Especially for screens of unbiased libraries, false positives can dominate "hit lists"; their origins are much debated. Here we determine the mechanism of every active hit from a screen of 70,563 unbiased molecules against beta-lactamase using quantitative HTS (qHTS). Of the 1,274 initial inhibitors, 95% were detergent-sensitive and were classified as aggregators. Among the 70 remaining were 25 potent, covalent-acting beta-lactams. Mass spectra, counter-screens, and crystallography identified 12 as promiscuous covalent inhibitors. The remaining 33 were either aggregators or irreproducible. No specific reversible inhibitors were found. We turned to molecular docking to prioritize molecules from the same library for testing at higher concentrations. Of 16 tested, 2 were modest inhibitors. Subsequent X-ray structures corresponded to the docking prediction. Analog synthesis improved affinity to 8 microM. These results suggest that it may be the physical behavior of organic molecules, not their reactivity, that accounts for most screening artifacts. Structure-based methods may prioritize weak-but-novel chemotypes in unbiased library screens.
|
|
Journal of Biomolecular Screening
|
Quantitative High Throughput Screening Using a Live Cell cAMP Assay Identifies Small Molecule Agonists of the TSH Receptor.
Titus S, Neumann S, Zheng W, Southall N, Michael S, Klumpp C, Shinn P, Thomas CJ, Inglese J, Gershengorn MC, Austin CP.
The thyroid stimulating hormone (TSH, thyrotropin) receptor belongs to the glycoprotein
hormone receptor subfamily of seven-transmembrane spanning receptors. TSH receptor (TSHR)
is expressed mainly in thyroid follicular cells and is activated by TSH, which regulates growth
and function of thyroid follicular cells. Recombinant TSH is used in diagnostic screens for
thyroid cancer, especially in patients after thyroid cancer surgery. Currently, no selective small
molecule agonists of the TSH receptor are available. To screen for novel TSH receptor agonists,
we miniaturized a cell-based cAMP assay into 1536-well plate format. This assay uses a
HEK293 cell line stably transfected with the TSHR coupled to a cyclic nucleotide gated ion
channel (CNG) as a biosensor. From a quantitative high-throughput screen of 73,180 compounds
in parallel with a parental cell line (without the TSHR), 276 primary active compounds were
identified. The activities of the selected active compounds were further confirmed in an
orthogonal HTRF cAMP-based assay. 49 compounds in several structural classes have been
confirmed as the small molecule TSHR agonists that will serve as starting point for chemical
optimization and studies of thyroid physiology in health and disease.
|
|
Journal of the Association for Laboratory Automation
|
Compound Management for Quantitative High-Throughput Screening.
Yasgar A, Shinn P, Jadhav A, Auld DS, Michael S, Zheng W, Austin CP, Inglese J, Simeonov A.
An efficient and versatile Compound Management operation is essential for the success
of all downstream processes in high-throughput screening (HTS) and small molecule lead
development. Staff, equipment, and processes need to be not only reliable, but remain flexible
and prepared to incorporate paradigm changes. In the present report, we describe a system and
associated processes which enable handling of compounds for both screening and follow-up
purposes at the NIH Chemical Genomics Center (NCGC), a recently-established HTS and probe
development center within the Molecular Libraries Initiative of the NIH Roadmap. Our
screening process, termed quantitative HTS (qHTS), involves assaying the complete compound
library, currently containing >200,000 members, at a series of dilutions to construct a full
concentration-response profile. As such, Compound Management at the NCGC has been
uniquely tasked to prepare, store, register, and track a vertically-developed plate dilution series
(i.e., inter-plate titrations) in the 384-well format. These are compressed into a series of 1,536-
well plates and are registered to track all subsequent plate storage. Here, we present details on
the selection of equipment to enable automated, reliable and parallel compound manipulation in
384- and 1,536-well formats, protocols for preparation of inter-plate dilution series for qHTS, as
well as qHTS-specific processes and issues.
|
|
ASSAY and Drug Development Technologies
|
Evaluation of Micro-Parallel Liquid Chromatography (uPLC) as a Method for HTS-coupled Actives Verification.
Simeonov A, Yasgar A, Klumpp C, Zheng W, Shafqat N, Oppermann U, Austin CP, Inglese J.
The identification of biologically active compounds from HTS can involve
considerable post-screening analysis to verify the nature of the sample activity. In this
study we evaluated the performance of Micro Parallel Liquid Chromatography (uPLC) as
a separation-based enzyme assay platform for follow-up of compound activities found in
qHTS of two different targets, a hydrolase and an oxidoreductase. In an effort to couple
secondary analysis to primary screening we explored the application of uPLC
immediately after a primary screen. In a proof-of-concept experiment for screen-coupled
actives verification, we identified, selected and consolidated the contents of "active"
wells from a 1536-well format HTS experiment into a 384-well plate, and subsequently
analyzed these samples by a 24-channel uPLC system. The method utilized 0.6% of the
original 6 uL 1536-well assay for the analysis. The analysis revealed several nonbiological
based "positive" samples. The main examples included "false" enzyme
activators resulting from an increase in well-fluorescence due to fluorescent compound or
impurity. The uPLC analysis also provided a verification of the activity of two activators
of glucocerebrosidase. We discuss the benefits of uPLC and its limitations from the
standpoint of ease of use and integration into a seamless post-screen workflow.
|
|
Environmental Health Perspectives
|
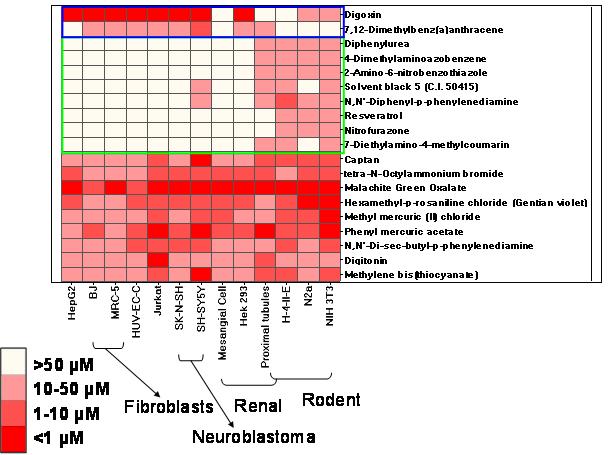
Compound Cytotoxicity Profiling Using Quantitative High-Throughput Screening.
Xia M, Huang R, Witt KL, Southall N, Fostel J, Cho MH, Jadhav A, Smith CS, Inglese J, Portier CJ, Tice RR, Austin CP.
Background: The propensity of compounds to produce adverse health effects in humans is
generally evaluated using animal-based test methods. Such methods can be relatively expensive,
low-throughput, and associated with pain suffered by the treated animals. In addition, differences
in species biology may confound extrapolation to human health effects.
Objective: The U.S. National Toxicology Program and the NIH Chemical Genomics Center are
collaborating to identify a battery of cell-based screens to prioritize compounds for further
toxicological evaluation.
Methods: 1,408 compounds previously tested in one or more traditional toxicological assays
were profiled for cytotoxicity using quantitative high-throughput screening (qHTS) in 13 human
and rodent cell types derived from six common targets of xenobiotic toxicity (liver, blood,
kidney, nerve, lung, skin). Selected cytotoxicants were further tested to define response kinetics.
Results: qHTS of these compounds produced robust and reproducible results which allowed
cross-compound, cross-cell type, and cross-species comparisons. Some compounds were
cytotoxic to all cell types at similar concentrations, while others exhibited species- or cell typespecific
cytotoxicity. Closely related cell types and analogous cell types in human and rodent
frequently showed different patterns of cytotoxicity. Some compounds inducing similar levels of
cytotoxicity showed distinct time-dependence in kinetic studies, consistent with known
mechanisms of toxicity.
Conclusions: The generation of high-quality cytotoxicity data on this large library of known
compounds using qHTS demonstrates the potential of this methodology to profile a much
broader array of assays and compounds, which, in aggregate, may be valuable for prioritizing
compounds for further toxicological evaluation, identifying compounds with particular
mechanisms of action, and potentially predicting in vivo biological response

|
|
Journal of the American Chemical Society
|
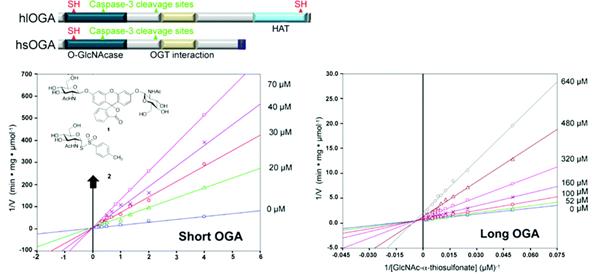
Distinctive Inhibition of O-GlcNAcase Isoforms by an alpha-GlcNAc Thiolsulfonate.
Kim, E. J.; Amorelli, B.; Abdo, M.; Thomas, C. J.; Love, D. C.; Knapp, S.; Hanover, J. A.
O-GlcNAcase (OGA) promotes O-GlcNAc removal, and thereby plays a key role in O-GlcNAc metabolism, a feature of a variety of vital cellular processes. Two splice transcripts of human OGA encode "long OGA", which contains a distinct N-terminal O-GlcNAcase domain and a C-terminal histoneacetylferase (HAT) domain, and "short OGA", which lacks the HAT domain. The functional roles of long OGA are only beginning to be unraveled, and the characteristics of short OGA remain almost unknown. We find that short OGA, which possesses O-GlcNAcase catalysis machinery like that of long OGA, exhibits comparative resistance to previously described potent inhibitors of long OGA and lysosomal hexosaminidases, including PUGNAc and NAG-thiazoline, suggesting a role for the HAT domain in O-GlcNAcase catalysis. We also find that -GlcNAc thiolsulfonate (2) is the most potent inhibitor of short OGA yet described (Ki = 10 M), and exhibits some degree of selectivity versus long OGA and lysosomal hexosaminidases. In contrast to its mode of inhibition of short OGA, 2 acts as a irreversible inhibitor of long OGA by covalently modifying the enzyme as an S-GlcNAc derivative. Covalent attachment of GlcNAc to the HAT domain of long OGA dramatically changes its properties with respect to enzymatic activity and caspase-3 cleavage.
|
|
Tetrahedron Letters
|
Synthesis of substituted 2-phenylhistamines via a microwave promoted Suzuki coupling.
Skoumbourdis, A. P., Moore, S., Landsman, M., Thomas, C. J.
Substitutions on the 2-position of the imidazole ring of histamine have proven useful in a number of biochemical settings. Current art for the synthesis of these constructs relies upon a cumbersome and low-yielding condensation reaction. Here-in we report a new procedure for the synthesis of a series of substituted 2-phenylhistamines utilizing a microwave-promoted Suzuki coupling.

|
|
Bioorganic and Medicinal Chemistry Letters
|
Identification of N-(quinolin-8-yl)benzenesulfonamides as agents capable of down-regulating NFkappaB activity within two separate high-throughput screens of NFkappaB activation.
Xie Y, Deng S, Thomas CJ, Liu Y, Zhang YQ, Rinderspacher A, Huang W, Gong G, Wyler M, Cayanis E, Aulner N, Többen U, Chung C, Pampou S, Southall N, Vidovic D, Schürer S, Branden L, Davis RE, Staudt LM, Inglese J, Austin CP, Landry DW, Smith DH, Auld DS.
We describe here a series of N-(quinolin-8-yl)benzenesulfonamides capable of suppressing the NFkappaB pathway identified from two high-throughput screens run at two centers of the NIH Molecular Libraries Initiative. These small molecules were confirmed in both primary and secondary assays of NFkappaB activation and expanded upon through analogue synthesis. The series exhibited potencies in the cell-based assays at as low as 0.6muM, and several indications suggest that the targeted activity lies within a common region of the NFkappaB pathway.

|
|
Bioorganic and Medicinal Chemistry Letters
|
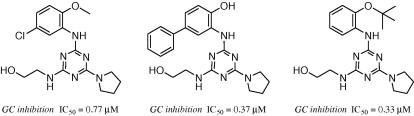
N4-phenyl modifications of N2-(2-hydroxyl)ethyl-6-(pyrrolidin-1-yl)-1,3,5-triazine-2,4-diamines enhance glucocerebrosidase inhibition by small molecules with potential as chemical chaperones for Gaucher disease.
Huang W, Zheng W, Urban DJ, Inglese J, Sidransky E, Austin CP, Thomas CJ.
A series of 1,3,5-triazine-2,4,6-triamines were prepared and analyzed as inhibitors of glucocerebrosidase. Synthesis, structure activity relationships and the selectivity of chosen analogues against related sugar hydrolases enzymes are described.
|
Neurobiology of Disease

|
Differentiating Alzheimer Disease-Associated Aggregates with Small Molecules
Honson NS, Johnson RL, Huang W, Inglese J, Austin CP, Kuret J.
Alzheimer disease is diagnosed postmortem by the density and spatial distribution of b-amyloid
plaques and tau-bearing neurofibrillary tangles. The major protein component of each lesion
adopts cross-b-sheet conformation capable of binding small molecules with submicromolar
affinity. In many cases, however, Alzheimer pathology overlaps with Lewy body disease,
characterized by the accumulation of a third cross-b-sheet forming protein, a-synuclein. To
determine the feasibility of distinguishing tau aggregates from b-amyloid and a-synuclein
aggregates with small molecule probes, a library containing 71,975 small molecules was
screened for antagonists of tau-aggregate mediated changes in Thioflavin S fluorescence,
followed by secondary screens to distinguish the relative affinity for each substrate protein.
Results showed that >10-fold binding selectivity among substrates could be achieved, with
molecules selective for tau aggregates containing at least three aromatic or rigid moieties
connected by two rotatable bonds.
|
Nature Chemical Biology

|
High-throughput screening assays for the identification of chemical probes
Inglese J, Johnson RL, Simeonov A, Xia M, Zheng W, Austin CP, Auld DS.
High-throughput screening (HTS) assays enable the testing of large numbers of chemical substances for activity in diverse areas of biology. The biological responses measured in HTS assays span isolated biochemical systems containing purified receptors or enzymes to signal transduction pathways and complex networks functioning in cellular environments. This Review addresses factors that need to be considered when implementing assays for HTS and is aimed particularly at investigators new to this field. We discuss assay design strategies, the major detection technologies and examples of HTS assays for common target classes, cellular pathways and simple cellular phenotypes. We conclude with special considerations for configuring sensitive, robust, informative and economically feasible HTS assays.

|
|
Nature Chemical Biology
|
Reporting data from high-throughput screening of small-molecule libraries
Inglese J, Shamu CE, Guy RK.
Publications reporting results of small-molecule screens are becoming more common as academic researchers increasingly make use of high-throughput screening (HTS) facilities. However, no standards have been formally established for reporting small-molecule screening data, and often key information important for the evaluation and interpretation of results is omitted in published HTS protocols. Here, we propose concise guidelines for reporting small-molecule HTS data.

|
European Pharmaceutical Review

|
HTS technologies to facilitate chemical genomics
Auld DS, Inglese J, Jadhav A, Austin CP, Sittampalam GS, Montrose-Rafizadeh C, Mcgee JE Iversen PW.
Industrial scale technologies developed and applied within the pharmaceutical industry for the purpose of drug discovery have recently been adopted by many research laboratories for the purpose of facilitating chemical genomics. Taking full advantage of these technologies will require education in highthroughput screening assay systems as well as new methods that exploit the capabilities of existing technologies.
|
|
Journal of Medicinal Chemistry
|
A high-throughput screen for aggregation-based inhibition in a large compound library.
Feng BY, Simeonov A, Jadhav A, Babaoglu K, Inglese J, Shoichet BK, Austin CP.
High-throughput screening (HTS) is the primary technique for new lead identification in drug discovery and chemical biology. Unfortunately, it is susceptible to false-positive hits. One common mechanism for such false-positives is the congregation of organic molecules into colloidal aggregates, which nonspecifically inhibit enzymes. To both evaluate the feasibility of large-scale identification of aggregate-based inhibition and quantify its prevalence among screening hits, we tested 70,563 molecules from the National Institutes of Health Chemical Genomics Center (NCGC) library for detergent-sensitive inhibition. Each molecule was screened in at least seven concentrations, such that dose-response curves were obtained for all molecules in the library. There were 1274 inhibitors identified in total, of which 1204 were unambiguously detergent-sensitive. We identified these as aggregate-based inhibitors. Thirty-one library molecules were independently purchased and retested in secondary low-throughput experiments; 29 of these were confirmed as either aggregators or nonaggregators, as appropriate. Finally, with the dose-response information collected for every compound, we could examine the correlation between aggregate-based inhibition and steep dose-response curves. Three key results emerge from this study: first, detergent-dependent identification of aggregate-based inhibition is feasible on the large scale. Second, 95% of the actives obtained in this screen are aggregate-based inhibitors. Third, aggregate-based inhibition is correlated with steep dose-response curves, although not absolutely. The results of this screen are being released publicly via the PubChem database.
|
|
ASSAY and Drug Development Technologies
|
A cell-based assay for IkappaBalpha stabilization using a two-color dual luciferase-based sensor.
Davis RE, Zhang YQ, Southall N, Staudt LM, Austin CP, Inglese J, Auld DS.
A cell-sensor assay for stabilization of IkappaBalpha was developed in the activated B cell-like diffuse large B-cell lymphoma cell line OCI-Ly3. This cell line expresses known nuclear factor kappaB (NFkappaB) target genes due to high constitutive activity of IkappaB kinase (IKK), which phosphorylates the protein IkappaBalpha leading to proteasomal degradation of IkappaBalpha and activation of NFkappaB. The cell-sensor assay uses green and red light-emitting beetle luciferases, with the green luciferase fused to IkappaBalpha (IkappaBalpha-CBG68) and the red luciferase (CBR) present in its native state. The IkappaBalpha-CBG68 reporter functions as a sensor of IKK and proteasome activity, while CBR serves to normalize for cell number and nonspecific effects. Both reporter constructs were stably integrated and placed under the control of an inducible promoter system, which increased fold responsiveness to inhibitors when assay incubations were performed simultaneous to reporter induction by doxycycline. The assay was miniaturized to a 1,536-well plate format and showed a Z' of 0.6; it was then used to panel 2,677 bioactive compounds by a concentration-response-based screening strategy. The concentration-effect curves for the IkappaBalpha-CBG68 and CBR signals were then used to identify specific stabilizers of IkappaBalpha, such as IKK inhibitors or proteasome inhibitors, which increased the doxycycline-induced rise in IkappaBalpha-CBG68 without affecting the rise in CBR. Known and unexpected inhibitors of NFkappaB signaling were identified from the bioactive collection. We describe here the development and performance of this assay, and discuss the merits of its specific features.

|
|
Methods in Enzymology
|
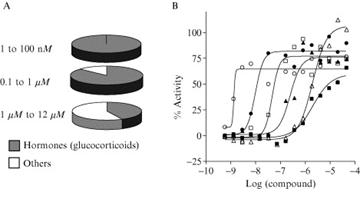
Fluorescent protein-based cellular assays analyzed by laser-scanning microplate cytometry in 1536-well plate format.
Auld DS, Johnson RL, Zhang YQ, Veith H, Jadhav A, Yasgar A, Simeonov A, Zheng W, Martinez ED, Westwick JK, Austin CP, Inglese J.
Microtiter plate readers have evolved from photomultiplier and charged-coupled device-based readers, where a population-averaged signal is detected from each well, to microscope-based imaging systems, where cellular characteristics from individual cells are measured. For these systems, speed and ease of data analysis are inversely proportional to the amount of data collected from each well. Microplate laser cytometry is a technology compatible with a 1536-well plate format and capable of population distribution analysis. Microplate cytometers such as the Acumen Explorer can monitor up to four fluorescent signals from single objects in microtiter plates with densities as high as 1536 wells. These instruments can measure changes in fluorescent protein expression, cell shape, or simple cellular redistribution events such as cytoplasmic to nuclear translocation. To develop high-throughput screening applications using laser-scanning microplate cytometry, we used green fluorescent protein- and yellow fluorescent protein-expressing cell lines designed to measure diverse biological functions such as nuclear translocation, epigenetic signaling, and G protein-coupled receptor activation. This chapter illustrates the application of microplate laser cytometry to these assays in a manner that is suitable for screening large compound collections in high throughput.
|
|
Drug Discovery Today
|
Measure, mine, model, and manipulate: the future for HTS and chemoinformatics?
Parker CN, Shamu CE, Kraybill B, Austin CP, Bajorath J.
|
Proceedings
of the National Academy of Sciences

|
Quantitative high-throughput screening: A titration-based approach that
efficiently identifies biological activities in large chemical libraries
Inglese J, Auld DS, Jadhav A, Johnson RL, Simeonov A, Yasgar A, Zheng W, Austin CP.
High-throughput screening (HTS) of chemical compounds to identify modulators of molecular targets is a mainstay of pharmaceutical development. Increasingly, HTS is being used to identify chemical probes of gene, pathway, and cell functions, with the ultimate goal of comprehensively delineating relationships between chemical structures and biological activities. Achieving this goal will require methodologies that efficiently generate pharmacological data from the primary screen and reliably profile the range of biological activities associated with large chemical libraries. Traditional HTS, which tests compounds at a single concentration, is not suited to this task, because HTS is burdened by frequent false positives and false negatives and requires extensive follow-up testing. We have developed a paradigm, quantitative HTS (qHTS), tested with the enzyme pyruvate kinase, to generate concentration-response curves for >60,000 compounds in a single experiment. We show that this method is precise, refractory to variations in sample preparation, and identifies compounds with a wide range of activities. Concentration-response curves were classified to rapidly identify pyruvate kinase activators and inhibitors with a variety of potencies and efficacies and elucidate structure-activity relationships directly from the primary screen. Comparison of qHTS with traditional single-concentration HTS revealed a high prevalence of false negatives in the single-point screen. This study demonstrates the feasibility of qHTS for accurately profiling every compound in large chemical libraries (>10(5) compounds). qHTS produces rich data sets that can be immediately mined for reliable biological activities, thereby providing a platform for chemical genomics and accelerating the identification of leads for drug discovery.
|
|
Drug Discovery Today
|
Expanding the HTS paradigm.
Inglese J.
|
|
Invited Lectures: (last updated August 2007)
-
59. Genetic Alliance Annual Conference: From Bench to Bedside to Practice: a practical course – moving toward treatment, Bethesda, MD, 27 July 2007, Bethesda North Marriott, Sponsored by the Office of Rare Diseases, NIH, DHHS
-
58. 50th Annual Meeting and Conference of the Canadian Society of Biochemistry, Molecular and Cellular Biology (CSBMCB), July 5- 9, 2007, McGill University, Montreal Quebec, Canada.
-
57. “Overview of the NIH Molecular Libraries Roadmap Initiative and the NCGC’s Quantitative High Throughput Screening Approach to Chemical Genomics”, Novel strategies for compound identification from compound libraries: High Throughput Screening, Biochemical Pharmacology Discussion Group, The New York Academy of Sciences, 7 World Trade Center – 40th floor, New York, NY, April 24, 2007.
-
56. “Quantitative High Throughput Screening: Improving Decision Making in Lead Identification by the Comprehensive Biological Activity Profiling of Chemical Libraries” ACS ProSpectives: Discovery & Selection of Successful Drug Candidates, Intercontinental Boston, Boston, MA, April 30, 2007
-
55. “Quantitative High Throughput Screening: Improving Decision Making in Lead Identification by the Biological Activity Profiling of Chemical Libraries”, High Throughput Screening Symposium, University of Kansas, Lawrence, KS, March 12, 2007, Chair: Jeff Aube
-
54. “Quantitative High Throughput Screening: Improving Decision Making in Lead Identification by the Biological Activity Profiling of Chemical Libraries”, PITTCON 2007 Conference and Expo, Drug Screening Assays: Opportunities for Analytical Chemistry in Drug Discovery, McCormick Place South , Chicago, IL, March 1, 2007
-
53. “qHTS and Novel Molecular Probes” NIH Chemistry Seminar Series, Hosted by the Chemistry Interest Group, National Institutes of Health, Building 50, Bethesda, MD, December 1, 2006
-
52. “Quantitative High Throughput Screening: Discovery of Investigational Molecular Probes though the Biological Activity Profiling of Chemical Libraries”, Assays & Cellular Targets 2006: High Content Analysis Conference, Green Valley Ranch Resort, Las Vegas, NV, October 31, 2006
-
51. “Quantitative High Throughput Screening: Discovery of Investigational Molecular Probes though the Biological Activity Profiling of Chemical Libraries”, Grand Challenges in Global Health 2nd Annual Meeting: High Throughput Screening and Chemical Libraries, Grand Hyatt Hotel, Washington, DC, October 5, 2006
-
50. “Quantitative High Throughput Screening: Discovery of Investigational Molecular Probes though the Biological Activity Profiling of Chemical Libraries”, Keck Seminar Series, Rice University, Houston, TX, September 29, 2006
-
49. “The NIH Molecular Libraries Initiative and the Molecular Libraries Screening Center Network”, Gordon Research Conference on Combinatorial Chemistry, The Queen’s College, Oxford, UK, 22 August 2006
-
48. “Quantitative High Throughput Screening: Discovery of Investigational Molecular Probes though the Biological Activity Profiling of Chemical Libraries”, Gordon Research Conference on Combinatorial Chemistry, The Queen’s College, Oxford, UK, 22 August 2006
-
47. “Quantitative High Throughput Screening: Discovery of Investigational Molecular Probes though the Biological Activity Profiling of Chemical Libraries”, Genetic Alliance Annual Conference: From Bench to Bedside to Practice: a practical course – moving toward treatment, Bethesda, MD, 28 July 2006
-
46. “Quantitative High Throughput Screening: Discovery of Investigational Molecular Probes though the Biological Activity Profiling of Chemical Libraries”, 7th Annual Symposium on Chemical Synthesis, Boston University, Boston, MA , 30 June 2006
-
45. “Quantitative High Throughput Screening: Discovery of Investigational Molecular Probes though the Biological Activity Profiling of Chemical Libraries”, IRIC - Institute for Research in Immunology and Cancer, Universite de Montreal, Montreal, Canada, 12 June 2006
-
44. “Quantitative High Throughput Screening: Discovery of Investigational Molecular Probes though the Biological Activity Profiling of Chemical Libraries”, International Society of Analytical Cytology XXIII International Congress, International Society of Analytical Cytology (ISAC), Quebec City, Canada , 21 May 2006
-
43. “Quantitative High Throughput Screening: Discovery of Investigational Molecular Probes though the Biological Activity Profiling of Chemical Libraries”, Harvard Chemical Biology Initiative, Harvard University, Department of Chemistry and Chemical Biology, Cambridge, MA, 18 May 2006
-
42. “Chemical Genomics 101”, National Toxicology Program, High Throughput Screening Initiative, National Institute of Environmental Health Sciences, Research Triangle, NC, 20 April 2006
-
41. “Frontiers of Screening: High Throughput Screening at the NCGC”, Pennsylvania Molecular Screening Summit. Hershey, PA, 13 April 2006
-
40. “Annotating the Biological Activity of Chemical Libraries using Quantitative High Throughput Screening”, SBS Regional Meeting. New Frontiers in Drug Discovery: Academic - Industry Synergies. Baltimore, MD, 3 April 2006
-
39. “Annotating the Biological Activity of Chemical Libraries using Quantitative High Throughput Screening”, Center for Chemical Genomics Colloquium. University of Michigan, 17 March 2006
-
38. “Annotating the Biological Activity of Chemical Libraries using Quantitative High Throughput Screening” Amgen, Thousand Oaks, CA, 27 February 2006
-
37. “Quantitative High Throughput Screening: Rethinking HTS for Chemical Genomics” National Toxicology Program, High Throughput Screening Assays Workshop, Crystal City, Arlington, VA, December 14-15, 2005
-
36. “Annotating the Biological Activity of Chemical Libraries using Quantitative High Throughput Screening”, Rensselaer Polytechnic Institute, Troy NY, 6-7 December 2005
-
35. “Annotating the Biological Activity of Chemical Libraries using Quantitative HTS”, AstraZeneca, Wilmington, DE, November 30, 2005.
-
34. “Annotating the Biological Activity of Chemical Libraries using Cellular Assays”, IBC’s Inaugural Conference on High Content Analysis, Washington, DC, November 18, 2005
-
33. “Annotating the Biological Activity of Chemical Libraries using Cellular Assays”, CHI’s Fluorescent Proteins For Cellular Imaging and Drug Development, La Jolla, CA, November 14-15, 2005
-
32. “Annotating the Biological Activity of Chemical Libraries and the Forging of Molecular Probes” NHGRI Faculty Seminar, Bethesda, MD, 18 October 2005.
-
31. “Profiling Signaling Pathways using Chemical Libraries” GE Healthcare Cellular Analysis Symposium, GE Global Research Center, Niskayuna, NY, 1 October 2005.
-
30. “Profiling Signaling Pathways using Chemical Libraries and ?-Lactamase Reporter Gene Technology”, SBS 11th Annual Conference & Exhibition Drug Discovery: From Targets to Candidates, Screen design and assay technology SIG, Geneva, Switzerland, 13 September 2005
-
29. “Profiling Phenotypic Assays using Chemical Libraries and Redistribution® Technology: Progress Report”, SBS 11th Annual Conference & Exhibition Drug Discovery: From Targets to Candidates, BioImage User's Meeting, Geneva, Switzerland, 12 September 2005
-
28. “Technologies Enabling the Mapping of Chemical Genomic Space”, SBS 11th Annual Conference & Exhibition Drug Discovery: From Targets to Candidates, Aurora Discovery User's Meeting, Geneva, Switzerland, 12 September 2005
-
27. “NIH Chemical Genomics Center: Profiling the Biological Activity of Novel Chemical Libraries” CMLD Program Meeting, Department of Molecular and Cellular Biology, Boston University, Boston, MA 02215, August 11, 2005.
-
26. “NIH Chemical Genomics Center: Opening a New Door to Discovery” Symposium on High Content Cellular Imaging. Department of Molecular and Cellular Biology, Baylor College of Medicine, Houston, TX. June 22, 2005.
-
25. “NIH Chemical Genomics Center: Opening a New Door to Discovery” Department of Pharmacology Seminar. University of North Carolina, Chapel Hill, NC. May 31, 2005. Host: Professor Henrik Dohlman
-
24. “NIH Chemical Genomics Center: Opening a New Door to Discovery” Plenary Talk for SBS Bay Area Regional Meeting Exploiting the Druggable Genome: A West Coast Focus. San Mateo Marriott, San Mateo, CA April 21 - 22, 2005.
-
23. “NIH Chemical Genomics Center: Opening a New Door to Discovery” FASEB Symposium. Chemical Genetics: Using Chemical Diversity to Understand Biological Mechanisms. San Diego Marriot Hotel and Marina, April 1, 2005.
-
22. “NIH Chemical Genomics Center: Opening a New Door to Discovery” Schering-Plough Research Institute (SPRI). Compound Repository Facility, Summit, NJ. February 10, 2005.
-
21. “Protein Domain-Based Biosensors: Applying Elements from Cellular Signaling to Assay Development and Drug Discovery” Society for Biomolecular Screening (SBS) 10th Anniversary Conference. Advances in Bioassay Technology, Orlando, FL., September 15, 2004.
-
20. “NIH Chemical Genomics Center: Opening a New Door to Discovery” Society for Biomolecular Screening (SBS) 10th Anniversary Conference. Academic Outreach Special Interest Group Meeting, Orlando FL., September 13, 2004.
-
19. “NIH Chemical Genomics Center: Opening a New Door to Discovery” Keynote address at the 12th Annual Conference of the Sino-American Pharmaceutical Professionals Association (SAPA). Busch Campus of Rutgers University, Piscataway, New Jersey, August 7, 2004.
-
18. “Automation and Miniaturization of Bioassays” 2003 Annual Meeting of the American Association of Pharmaceutical Scientists (AAPS). Salt Lake City, Utah, October 26-30, 2003.
-
17. “Assay Miniaturization and Automation for Drug Discovery” National Human Genomics Research Institute, NIH Building 31, Bethesda, MD, September 24, 2003.
-
16. “Assay Miniaturization and Automation for Drug Discovery” Vanderbilt Institute of Chemical Biology, Vanderbilt University, Nashville, TN, July 28, 2003.
-
15. “Imaging Microscopy: Systems and methods for the analysis of cellular assays and micro-fabricated arrays and chips” Merck Technology Symposium 2003, PineCrest Country Club, Montgomeryville, PA. May 20th 2003.
-
14. “PDZ Domain-Based Product Sensors: Applying Elements from Cellular Signaling to Assay Development and Drug Discovery” IBC’s 6th Annual Conference on Assay Development, San Diego, CA, October 16-18, 2002.
-
13. “Assay Miniaturization and Automation” IBC’s 5th Annual Conference on Assay Development: Developing Robust Assays for Screening through Advanced Technologies, Coronado, CA, November 28-30, 2001
-
12. “Lead Discovery Using Encoded Combinatorial Libraries” American Chemical Society 216th National Meeting, Boston, MA, August 23rd, 1998.
-
11. “Chemokine Receptor-Ligand Interactions Measured Using Time-Resolved Fluorescence”, IBC’s Fifth Industry Symposium on Chemokines, San Francisco, CA, September 24-25, 1998.
-
10. "Strategies for Screening Signal Transduction Targets with Combinatorial Libraries", IBC's Third Annual Conference on High-Throughput Screening: Novel Assay Design for Transduction / Transcription Based Drug Discovery, San Diego, CA, September 23, 1997.
-
9. "Screening of Signal Transduction Targets with Combinatorial Libraries", IBC's Second Annual Conference on High-Throughput Screening: Novel Assay Design for Transduction / Transcription Based Drug Discovery, San Diego, CA, September 18, 1996.
-
8. "Lipid Modifications of G Protein-Coupled Receptor Kinases", The 658th Biochemical Society Meeting, University of Liverpool, UK, April 19, 1996.
-
7. "A Novel Family of Protein Kinases that Regulate G Protein-Coupled Receptor Signal Transduction", Prof. H. G. Khorana’s Group, Department of Biology & Chemistry, Massachusetts Institute of Technology, January 20, 1994.
-
6. "Regulation of Serpentine Receptor Signaling by a Novel Family of Protein Kinases", Department of Pharmacology, University of Washington, Seattle, WA, October 19, 1994.
-
5. "Isoprenylation in the Regulation of Sensory Receptors", 25th Annual Meeting of the American Society for Neurochemistry, Albuquerque, NM, March 5-9, 1994.
-
4. "Isoprenylation in the Regulation of G Protein-Coupled Receptors", The 75th Annual Meeting of the Endocrine Society, Las Vegas, NV, June 9-12, 1993.
-
3. "Isoprenylation in the Regulation of G Protein-Coupled Signal Transduction", Berman-Gund Laboratory, Massachusetts Eye & Ear Infirmary, Harvard Medical School, Boston, MA, January 22, 1993.
-
2. "Mechanisms of G Protein-Coupled Receptor Regulation", Organized by the Duke University Cell and Molecular Biology Departments and the Duke Comprehensive Cancer Center, Spring Symposium on "Signal Transduction", May 2, 1992.
-
1. "Regulation of G Protein-Coupled Receptors: The Receptor Kinase Family", Bio Japan '92, Organized by the Japan Bioindustry Association (JBA), Pacific Convention Plaza Yokohama, Japan, August 26-28, 1992.
|
|






 (301) 217-5723
(301) 217-5723 (301) 217-5736
(301) 217-5736