Reviewed February 2007
What is the official name of the HBB gene?
The official name of this gene is “hemoglobin, beta.”
HBB is the gene's official symbol. The HBB gene is also known by other names, listed below.
What is the normal function of the HBB gene?
The HBB gene provides instructions for making a protein called beta hemoglobin. Beta hemoglobin is a subunit of a larger protein called hemoglobin, which is located inside red blood cells. Hemoglobin normally consists of four protein subunits: two subunits called beta hemoglobin and two subunits called alpha hemoglobin. The instructions for alpha hemoglobin are carried in a separate gene.
Each protein subunit of hemoglobin carries an iron-containing molecule called heme. Heme molecules are necessary for red blood cells to pick up oxygen in the lungs and deliver it throughout the body. A complete hemoglobin protein is capable of carrying four oxygen molecules at a time. Oxygen binding to hemoglobin gives blood its bright red color.
How are changes in the HBB gene related to health conditions?
-
beta thalassemia - caused by mutations in the HBB gene
-
More than 200 HBB mutations that cause beta thalassemia have been identified. Most of the mutations involve a change in a single DNA building block (nucleotide) within or near the HBB gene. Other mutations insert or delete a small number of nucleotides in the HBB gene.
HBB mutations that decrease beta hemoglobin production result in a condition called beta-plus (B+) thalassemia. Mutations that prevent cells from producing any beta hemoglobin result in beta-zero (B0) thalassemia. Without proper amounts of beta hemoglobin, red blood cells cannot bind enough oxygen to satisfy the body's needs.
-
methemoglobinemia, beta-globin type - caused by mutations in the HBB gene
-
Mutations in specific regions of the HBB gene cause red blood cells to produce an abnormal form of hemoglobin called hemoglobin M. This form of hemoglobin disrupts the protein's interaction with iron and interferes with the delivery of oxygen to cells. As a result, people with this condition may have a bluish cast to their skin, mucous membranes, and underneath their fingernails.
-
sickle cell disease - caused by mutations in the HBB gene
-
Sickle cell anemia, a common form of sickle cell disease, is caused by a particular mutation in the HBB gene. This mutation results in the production of an abnormal version of beta hemoglobin called hemoglobin S or HbS. In this condition, hemoglobin S replaces both beta hemoglobin subunits in hemoglobin. The mutation changes a single protein building block (amino acid) in beta hemoglobin. Specifically, the amino acid glutamic acid is replaced with the amino acid valine at position 6 in beta hemoglobin, written as Glu6Val or E6V. Replacing glutamic acid with valine causes the abnormal HbS subunits to stick together and form long, rigid molecules. The rigid HbS molecules bend red blood cells into a sickle (crescent) shape. The sickle-shaped cells die prematurely, which can lead to a shortage of red blood cells (anemia). The sickle-shaped cells can also block small blood vessels, causing pain and organ damage.
Mutations in the HBB gene can also cause other abnormalities in beta hemoglobin. These abnormal forms of hemoglobin are often designated by letters of the alphabet and sometimes also by a name. In these other types of sickle cell disease, just one beta hemoglobin subunit is replaced with hemoglobin S. The other beta hemoglobin subunit is replaced with a different abnormal variant, such as hemoglobin C or hemoglobin E. One such condition is hemoglobin SC (HbSC) disease, in which the beta hemoglobin subunits are replaced by hemoglobin S and hemoglobin C. The severity of this disorder is quite variable, but it can be as severe as sickle cell anemia. Hemoglobin E (HbE) is a variant of hemoglobin most commonly found in the people of Southeast Asia. In some cases, the hemoglobin E mutation is present with either beta thalassemia or hemoglobin S. In these cases, a person may have more severe signs and symptoms associated with these disorders, such as episodes of pain, anemia, and abnormal spleen function.
Other conditions, known as hemoglobin sickle-beta thalassemias (HbSBetaThal), are caused when mutations that produce hemoglobin S and beta thalassemia occur together. The signs and symptoms of hemoglobin S-beta thalassemias are usually more severe than those of hemoglobin SC disease, and may include severe pain and organ damage.
- other disorders - caused by mutations in the HBB gene
-
Hundreds of alterations have been identified in the HBB gene. These changes result in the production of different versions of beta hemoglobin. Some of these variations have no noticeable signs or symptoms, while others may affect a person's health. Two of the most common variants are hemoglobin C and hemoglobin E.
Hemoglobin C (HbC) is more common in people of West African descent than in other populations. Hemoglobin C results when the amino acid lysine replaces the amino acid glutamic acid at position 6 in the beta hemoglobin subunit (written as Glu6Lys). People who have two hemoglobin C subunits in their hemoglobin, instead of beta hemoglobin, have a mild condition called hemoglobin C disease. This condition often causes chronic anemia in which the red blood cells are broken down prematurely.
Hemoglobin E (HbE) is a variant of hemoglobin most commonly found in the people of Southeast Asia. The mutation responsible for producing hemoglobin E substitutes the amino acid lysine for the amino acid glutamic acid at position 26 of beta hemoglobin (written as Glu26Lys). When a person has two hemoglobin E subunits in their hemoglobin, instead of beta hemoglobin, a mild anemia called hemoglobin E disease can occur.
Where is the HBB gene located?
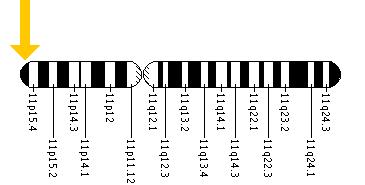
Cytogenetic Location: 11p15.5
Molecular Location on chromosome 11: base pairs 5,203,271 to 5,204,876
The HBB gene is located on the short (p) arm of chromosome 11 at position 15.5.
More precisely, the HBB gene is located from base pair 5,203,271 to base pair 5,204,876 on chromosome 11.
See How do geneticists indicate the location of a gene? in the Handbook.
Where can I find additional information about HBB?
You and your healthcare professional may find the following resources about HBB helpful.
- MedlinePlus - Health information
-
-
-
Gene Tests - DNA tests ordered by healthcare professionals (6 links)
You may also be interested in these resources, which are designed for genetics professionals and researchers.
- PubMed
 - Recent literature
- Recent literature
- OMIM - Genetic disorder catalog
-
What other names do people use for the HBB gene or gene products?
- beta globin
- HBB_HUMAN
- hemoglobin--beta locus
Where can I find general information about genes?
The Handbook provides basic information about genetics in clear language.
These links provide additional genetics resources that may be useful.
What glossary definitions help with understanding HBB?
The resources on this site should not be used as a substitute for
professional medical care or advice. Users seeking information about
a personal genetic disease, syndrome, or condition should consult with a qualified
healthcare professional.
See How can I find a genetics professional in my area? in the Handbook.