

GROMACS (www.gromacs.org) is a versatile package to perform molecular dynamics, i.e. simulate the Newtonian equations of motion for systems with hundreds to millions of particles. It is primarily designed for biochemical molecules like proteins and lipids that have a lot of complicated bonded interactions, but since GROMACS is extremely fast at calculating the nonbonded interactions (that usually dominate simulations) many groups are also using it for research on non-biological systems, e.g. polymers.
GROMACS manual, downloadable in several formats.
Versions
| Gromacs Version | To be used on | Add this to path | Parallel executable |
| 3.3.3 | all gige nodes | /usr/local/gromacs/bin /usr/local/mpich/bin |
mdrun_mpi |
| 3.3.3 | IB nodes | /usr/local/gromacs/bin-ib | mdrun_mpi |
| 3.3.1 | all gige nodes | /usr/local/gromacs-3.3.1/bin /usr/local/mpich/bin |
mdrun_mpi |
| 3.3.1 | Myrinet | /usr/local/gromacs-3.3.1/bin /usr/local/mpich-gm2k/bin |
mdrun_mpi_myr |
| 3.3.1 | IB nodes | /usr/local/gromacs-3.3.1/bin-ib | mdrun_mpi |
For basic information about setting up GROMACS jobs, read the GROMACS documentation. A collection of sample jobs is in /usr/local/gromacs/share/tutor.
All Biowulf nodes have 2 processors on each node. For greatest efficiency, GROMACS jobs should be run on both processors of each node. i.e. the job should be submitted with the -np=# parameter (no. of processors) equal to twice the -nodes=# parameter (no. of nodes). Note that non-Myrinet Gromacs does not scale beyond 2 processors, or 1 node.
Sample script for a GROMACS run:
-----------------------------------------------------
#!/bin/csh
# This is file Run_Gromacs
#PBS -N GROMACS
#PBS -k oe
#PBS -m be
#
set path = (/usr/local/mpich/bin /usr/local/gromacs/bin $path .)
cd /data/username/my_gromacs_runs/xyz/
grompp -np $np -shuffle -f md -c cpeptide_b4md -p cpeptide \
-o cpeptide_md >& ! out.run
mpirun -machinefile $PBS_NODEFILE -np $np /usr/local/gromacs/bin/mdrun_mpi \
-np $np -s cpeptide_md -o cpeptide_md -c cpeptide_after_md -v >>& ! out.run
------------------------------------------------------
Note that you need the '-np $np' flag in two places on the last line; one for the mpirun command and one for the mdrun program. This job would be submitted with:
qsub -v np=2 -l nodes=1:p1400 Run_Gromacs
Before submitting jobs to the Infinipath nodes, you must copy a file to your ~/.ssh directory:
% cd ~/.ssh(If you get an error that the .ssh directory does not exist, type mkdir ~/.ssh to create the directory)
% cp /usr/local/etc/ssh_config_ib config % chmod 600 config(If you already have a ssh config file, you should append the contents of /usr/local/etc/ssh_config_ib to it). This ssh config needs to be done only once, before submitting your first IB job.
Sample script for a Gromacs job submitted to the Infinipath/Opteron nodes:
#!/bin/csh
# This is file Run_Gromacs
#PBS -N Gromacs
#PBS -k oe
#PBS -m be
#
set path = (/usr/local/gromacs/bin-ib /usr/local/bin $path .)
cd /data/user/my_gromacs_dir
grompp -np $np -shuffle -sort -f Grompp.mdp -c Conf.gro -p Topol.top \
-o topol.tpr >>&! output
mpirun -machinefile $PBS_NODEFILE -np $np /usr/local/gromacs/bin-ib/mdrun_mpi \
-np $np -s topol.tpr -o traj.trr -c out.after_md -v >>&! output
This script would be submitted with:
qsub -v np=16 -l nodes=8:ipath Run_Gromacs
All Biowulf nodes have 2 processors on each node. For greatest efficiency, GROMACS jobs should be run on both processors of each node. i.e. the job should be submitted with the -np=# parameter (no. of processors) equal to twice the -nodes=# parameter (no. of nodes). Sample script for a Myrinet GROMACS run:
#!/bin/csh
# This is file Run_Gromacs
#PBS -N GROMACS
#PBS -k oe
#PBS -m be
#
set path = (/usr/local/mpich-gm2k/bin /usr/local/gromacs-3.3.1/bin /usr/local/bin $path .)
cd $PBS_O_WORKDIR
grompp -np $np -shuffle -sort -f Grompp.mdp -c Conf.gro -p Topol.top \
-o topol.tpr >>&! output1
mpirun -machinefile $PBS_NODEFILE -np $np /usr/local/gromacs-3.3.1/bin/mdrun_mpi_myr \
-np $np -s topol.tpr -o traj.trr -c out.after_md -v >>&! output1
This job would be submitted with:
qsub -v np=4 -l nodes=2:o2200:myr2k Run_Gromacs
It is critical to determine the appropriate number of nodes on which to run your job. As shown in the benchmarks below, different jobs scale differently. Thus, one job which scales very well could be submitted on up to 10 nodes, while another job may scale only up to 2 nodes. For some jobs, if you submit to more nodes than is optimal, your job will actually run slower. (e.g. the d.villin benchmark runs slower on 6 Myrinet nodes than on 2 Myrinet nodes).
To determine the optimal number of nodes:
- Set up a small version of your job. e.g. if your project involves a simulation of 100 ps, set up a 1ps job.
- Submit this job to 2 processors (1 node), 4processors (2 nodes), 6 processors (3 nodes) ...
- Examine the results. You want to pick the number of processors with at
least 60% efficiency.
100 * Time on 1 processor --------------------------- = Efficiency n * Time on n processors
Monitoring your jobs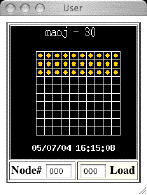 You can
use the Biowulf system monitors to watch your job.
Click on 'List status of Running Jobs only', and then on your username in the
resultant web page. This will pop up a small window showing the status of all
nodes that are running your jobs, as in the picture on the right. In the ideal
situation, all your nodes should be yellow (both processors used). Watch for
green or red nodes. Clicking on a node will pop up a window with information
about the processes running on that node.
You can
use the Biowulf system monitors to watch your job.
Click on 'List status of Running Jobs only', and then on your username in the
resultant web page. This will pop up a small window showing the status of all
nodes that are running your jobs, as in the picture on the right. In the ideal
situation, all your nodes should be yellow (both processors used). Watch for
green or red nodes. Clicking on a node will pop up a window with information
about the processes running on that node.The PBS batch system will write 2 files called JOBNAME.oJOBNUM and JOBNAME.eJOBNUM in your home directory (e.g. GROMACS.o90763 and GROMACS.e90763). They contain the standard output and standard error from your job. Most problems can be figured out by examining these files.
BenchmarksSummary:
- Different Gromacs jobs scale differently; it is important to run your own benchmarks to determine the optimal number of nodes for your particular job.
- Gromacs scales best on the Infiniband Opteron nodes. Based on these results, one could submit an 16 p (8 node) or 24p (12 node) Gromacs job to the IB nodes.
- The non-myrinet MPI version of Gromacs does not scale beyond 2 nodes (4 processors).
Benchmark 1: The DPPC membrane system from the Gromacs benchmark suite. Detailed results

