What Is Cystic Fibrosis?
Cystic fibrosis (CF) is an inherited disease of your
mucus and sweat glands. It affects mostly your lungs, pancreas, liver,
intestines, sinuses, and sex organs.
Normally, mucus is watery. It keeps the linings of
certain organs moist and prevents them from drying out or getting infected. But
in CF, an abnormal gene causes mucus to become thick and sticky.
The mucus builds up in your lungs and blocks the
airways. This makes it easy for bacteria to grow and leads to repeated serious
lung infections. Over time, these infections can cause serious damage to your
lungs.
The thick, sticky mucus can also block tubes, or
ducts, in your pancreas. As a result, digestive enzymes that are produced by
your pancreas cannot reach your small intestine. These enzymes help break down
the food that you eat. Without them, your intestines cannot absorb fats and
proteins fully.
As a result:
- Nutrients leave your body unused, and you can
become malnourished.
- Your stools become bulky.
- You may not get enough vitamins A, D, E, and
K.
- You may have intestinal gas, a swollen belly, and
pain or discomfort.
The abnormal gene also causes your sweat to become
extremely salty. As a result, when you perspire, your body loses large amounts
of salt. This can upset the balance of minerals in your blood. The imbalance
may cause you to have a heat emergency.
CF can also cause infertility (mostly in men).
The symptoms and severity of CF vary from person to
person. Some people with CF have serious lung and digestive problems. Other
people have more mild disease that doesn't show up until they are adolescents
or young adults.
Respiratory failure is the most common cause of
death in people with CF.
Until the 1980s, most deaths from CF occurred in
children and teenagers. Today, with improved treatments, people with CF live,
on average, to be more than 35 years old. Research continues to look for:
Other Names for Cystic Fibrosis
- CF
- Cystic fibrosis of the pancreas
- Fibrocystic disease of the pancreas
- Mucoviscidosis
- Mucoviscidosis of the pancreas
- Pancreas fibrocystic disease
- Pancreatic cystic fibrosis
What Causes Cystic Fibrosis?
Cystic fibrosis (CF) is caused by a defect in a gene
called the cystic fibrosis transmembrane conductance regulator (CFTR) gene.
This gene makes a protein that controls the movement of salt and water in and
out of the cells in your body. In people with CF, the gene does not work
effectively. This causes the thick, sticky mucus and very salty sweat that are
the main features of CF.
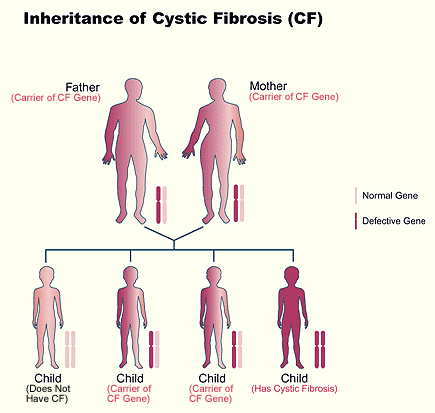
Each of us inherits two CFTR genes, one from each
parent.
- Children who inherit an abnormal CFTR gene from
each parent will have CF.
- Children who inherit an abnormal CFTR gene from
one parent and a normal CFTR gene from the other parent will not have CF. They
will be CF carriers.
CF carriers:
- Usually have no symptoms of CF
- Live normal lives
- Can pass the abnormal CFTR gene on to their
children
When two CF carriers have a baby, the baby has a:
- One in four chance of inheriting two abnormal
CFTR genes and having CF.
- One in four chance of inheriting two normal CFTR
genes and not having CF or being a carrier.
- Two in four chance of inheriting one normal CFTR
gene and one abnormal CFTR gene. The baby will not have CF but will be a CF
carrier like its parents.
Who Is At Risk for Cystic Fibrosis
About 30,000 people in the United States have cystic
fibrosis (CF).
- It affects both males and females.
- It affects people from all racial and ethnic
groups but is most common among Caucasians whose ancestors came from northern
Europe.
CF is one of the most common inherited diseases
among Caucasians.
About 1 in every 3,000 babies born in the United
States has CF.
CF is also common in:
- Latinos
- Native Americans, especially the Pueblo and Zuni
CF is much less common among:
- African Americans
- Asian Americans
About 12 million Americans are carriers of an
abnormal CF gene. Many of them do not know that they are CF carriers.
What Are the Signs and Symptoms of Cystic
Fibrosis?
Most of the symptoms of cystic fibrosis (CF) are
caused by the thick, sticky mucus. The most common symptoms include:
- Frequent coughing that brings up thick sputum, or
phlegm (flem).
- Frequent bouts of bronchitis and pneumonia. They
can lead to inflammation and permanent lung damage.
- Salty-tasting skin.
- Dehydration.
- Infertility (mostly in men).
- Ongoing diarrhea or bulky, foul-smelling, and
greasy stools.
- Huge appetite but poor weight gain and growth.
This is called "failure to thrive." It is a result of chronic malnutrition
because you do not get enough nutrients from your food.
- Stomach pain and discomfort caused by too much
gas in your intestines.
CF can also lead to other medical problems,
including:
- Sinusitis. The sinuses are air-filled spaces behind your eyes,
nose, and forehead. They produce mucus and help keep the lining of your nose
moist. When the sinuses become swollen, they get blocked with mucus and can
become infected. Most people with CF develop sinusitis.
- Bronchiectasis. Bronchiectasis is a lung
disease in which the bronchial tubes, or large airways in your lungs, become
stretched out and flabby over time and form pockets where mucus collects. The
mucus provides a breeding ground for bacteria. This leads to repeated lung
infections. Each infection does more damage to the bronchial tubes. If not
treated, bronchiectasis can lead to serious illness, including respiratory
failure.
- Pancreatitis. Pancreatitis is inflammation in the
pancreas that causes pain.
- Episodes of intestinal blockage, especially in
newborns.
- Nasal polyps, or growths in your nose, that may
require surgery.
- Clubbing. Clubbing is the widening and rounding
of the tips of your fingers and toes. It develops because your lungs are not
moving enough oxygen into your blood stream.
- Collapsed lung. This is also called
pneumothorax.
- Rectal prolapse. Frequent coughing or problems
passing stools may cause rectal tissue from inside you to move out of your
rectum.
- Liver disease due to inflammation or blocked bile ducts.
- Diabetes.
- Gallstones.
- Low bone density because you do not get enough
Vitamin D.
How Is Cystic Fibrosis Diagnosed?
First, your doctor will obtain a detailed medical
and family history and perform a thorough physical examination. Next, your
doctor may order some tests to ensure an accurate diagnosis.
The
sweat test is the most useful test for diagnosing cystic
fibrosis (CF). It measures the amount of salt in your sweat. For this test,
doctors rub a small amount of a chemical called pilocarpine (pi-lo-KAR-pen) on
your arm or leg. They then attach an electrode to this spot. The electrode
provides a mild electric current that produces sweat. This may cause tingling
or a feeling of warmth. They then cover the area with a gauze pad or filter
paper and wrap in plastic. After 30 to 40 minutes, they remove the plastic so
the sweat that collected on the pad or paper can be analyzed. The test is
usually done twice. High salt levels mean CF.
Your doctor may also do the following tests to
understand more about your condition and how to treat it:
- Blood tests to look for an abnormal CF gene or
other things that indicate CF.
- Chest x ray. A chest x ray takes a picture of your lungs. It
can show scarring from inflammation in your lungs.
- Sinus x ray. This test may show signs of sinusitis.
- Lung function tests can measure:
- How much air your lungs can hold
- How quickly you can breathe air out of your
lungs
- How well your lungs add oxygen to and remove
carbon dioxide from your blood
- Sputum (phlegm) cultures. Doctors take a sample
of your sputum to see what bacteria are growing in it.
If you are pregnant, prenatal genetic tests can find
out if your baby has CF:
- In
amniocentesis (AM-ne-o-sen-TE-sis), your doctor inserts a
hollow needle through your abdominal wall into your uterus to obtain cells from
the fluid (amniotic fluid) around the baby. The fluid is then tested to see if
both of the baby's CFTR genes are normal.
- In a
chorionic villus biopsy (ko-re-ON-ik VIL-us BI-op-se), your
doctor uses
ultrasound to guide a thin tube through your vagina and cervix
into your uterus and remove a tiny piece of the placenta to biopsy. The cells
of the placenta are then tested to see if the baby has CF.
Some States are now testing the blood of all
newborns for CF.
CF Carrier Testing
You may want to check whether you are a CF carrier,
if:
- You have a family history of CF.
- You are a partner of someone with CF.
- You are a couple planning a pregnancy.
A genetics counselor at your local hospital can take
a blood or saliva sample to see if it contains the abnormal CFTR gene that
causes CF. It will detect 9 out of 10 cases of CF. Some insurance plans cover
genetic testing.
How Is Cystic Fibrosis Treated?
There still is no cure for cystic fibrosis (CF), but
treatments have improved greatly in recent years. The goals of CF treatment are
to:
- Prevent and control infections in your
lungs.
- Loosen and remove the thick, sticky mucus from
your lungs.
- Prevent blockages in your intestines.
- Provide adequate nutrition.
Treatment for Lung Problems
The main treatments for lung problems in people with
CF are:
- Antibiotics for infections of the airways
- Chest physical therapy
- Exercise
- Other medications
Antibiotics
Most people with CF have ongoing, low-grade lung
infections. Sometimes, these infections become so serious that you may need to
be hospitalized. Antibiotics are the primary treatment.
You may be given several different types of
antibiotics. The choice of antibiotics depends on:
- The strains of bacteria involved
- How serious your condition is
- Your previous history of antibiotic use
The different types of antibiotics include:
- Oral antibiotics for relatively mild airway
infections.
- Inhaled antibiotics, such as tobramycin
(to-bra-MI-sin). They may be used alone or with oral antibiotics.
- Intravenous antibiotics for severe infections or
when none of the oral antibiotics work.
- Antibiotics, such as azithromycin
(az-ith-roe-MYE-sin), that also reduce inflammation.
Chest physical therapy
Chest physical therapy (CPT) is also called chest
clapping or percussion. It involves pounding your chest and back over and over
again to dislodge the mucus from your lungs so that you can cough up the mucus.
CPT for cystic fibrosis should be done three to four times each day.
CPT is also often referred to as postural drainage.
This involves your sitting or lying on your stomach with your head down while
you do CPT. This allows gravity to help drain the mucus from your lungs.
Because CPT is hard or uncomfortable for some
people, several devices have been developed recently that may help with CPT.
The devices include:
- An electric chest clapper, known as a mechanical
percussor.
- An inflatable therapy vest that uses
high-frequency air waves to force the mucus out of your lungs.
- A "flutter" device, a small hand-held device that
you breathe out through. It causes vibrations that dislodge the mucus.
- A positive expiratory pressure (PEP) mask that
creates vibrations that help break the mucus loose from the airway walls.
Several breathing techniques may also help dislodge
the mucus. These techniques include:
- Forced expiration technique (FET)—forcing
out a couple of breaths or huffs and then doing relaxed breathing
- Active cycle breathing (ACB)—FET with deep
breathing exercises that can loosen the mucus in your lungs and help open your
airways
Exercise
Aerobic exercise helps:
- Loosen the mucus
- Encourage coughing to clear the mucus
- Improve your overall physical condition
If you exercise regularly, you may be able to cut
back on your chest therapy. Check with your doctor before doing this.
Other medications
Anti-inflammatory medications may help reduce the
inflammation in your lungs that is caused by ongoing infections. These
medications include:
- Inhaled or, sometimes, oral steroids. Steroids
are the most effective anti-inflammatory medicines.
- Ibuprofen, a type of nonsteroidal,
anti-inflammatory medicine. It may slow the progress of CF in young children
with mild symptoms.
- Bronchodilators, which are inhaled drugs that
relax the muscles around the airways so that the airways can open up. They
should be taken just before CPT to help clear mucus.
Mucus-thinning drugs reduce the stickiness of mucus
in your airways. They include:
- Human DNase (Dornase Alfa), a drug that loosens
the mucus in your lungs. It may lead to shorter hospital stays.
- Acetylcysteine and saline.
- Hypertonic saline, a solution of very salty
sterile water taken by nebulizer two times a day, can help clear mucus and
improve lung function. Some doctors are now giving it to select patients over 6
years old.
Oxygen Therapy
If the level of oxygen in your blood is too low, you
may need oxygen therapy. Oxygen is usually given through nasal prongs or a
mask.
Lung Transplantation
Surgery to replace one or both of your lungs with
healthy lungs from a human donor may help you. Some of the factors that
determine whether you can undergo
lung transplantation include:
- The type of bacteria in your lungs
- Your age and weight
- The medications you are taking
- Whether you have other medical conditions,
including osteoporosis
- How well your lungs are functioning
Management of Digestive Problems
Nutritional therapy can improve your growth and
development, strength, and exercise tolerance. It may also make you strong
enough to resist some lung infections. Nutritional therapy includes a
well-balanced diet that is rich in calories, fat, and protein.
As part of your nutritional therapy, your doctor
may:
- Prescribe oral pancreatic enzymes to help you
digest fats and proteins and absorb more vitamins. The enzymes should be taken
in capsule form before every meal, including snacks.
- Recommend supplements of vitamins A, D, E, and K
to replace the fat-soluble vitamins that your intestines cannot absorb.
- Recommend that you use a feeding tube, called a
gastrostomy (gas-TROS-to-me) tube or T-tube, to add more calories at night
while you are sleeping. The tube is placed in your stomach. Before you go to
bed each night, you attach a bottle with a nutritional solution to the entrance
of the tube. It feeds you while you sleep.
Other treatments for the digestive problems caused
by CF may include:
- Enemas and mucus-thinning medications to treat
intestinal blockages
- Medicines that reduce stomach acid and help the
oral pancreatic enzymes work better
Living With Cystic Fibrosis
If you have cystic fibrosis (CF), you should learn
as much as you can about the disease and work closely with your doctors to
learn how to manage it.
Ongoing medical care is important. You should seek
treatment from a team of doctors, nurses, and respiratory therapists who
specialize in CF. These specialists are often located at
CF Foundation Centers in major medical centers.
Good self-care includes:
- Eating a healthy diet
- Avoiding tobacco smoke
- Washing your hands often to reduce your chances
of infection
- Exercising frequently
- Drinking lots of fluids
- Doing chest physical therapy every day
- Having annual flu and other appropriate
vaccinations
- Taking your medicines as prescribed
You can expect to have a normal sex life.
- Most men with CF are infertile, but they may be
helped with modern reproductive techniques.
- Although most women with CF may be less fertile
than women who don’t have CF, they usually can have children. Talk to
your doctor before becoming pregnant.
Having a positive attitude is also helpful.
If you are a parent of someone with CF, do not feel
guilty about passing it on to your child. And do not be overprotective;
encourage your child to be active and self-reliant.
Key Points
- Cystic fibrosis (CF) is an inherited disease of
your mucus and sweat glands. It affects mostly your lungs, pancreas, liver,
intestines, sinuses, and sex organs.
- In CF, an abnormal gene called the cystic
fibrosis transmembrane conductance regulator (CFTR) gene causes mucus to become
thick and sticky. The mucus builds up in the lungs and blocks the airways,
creating an environment that makes it easy for bacteria to grow. This leads to
repeated serious lung infections that can damage your lungs.
- The mucus can also block tubes, or ducts, in your
pancreas so that the digestive enzymes it produces cannot reach the intestines
where they are needed to break down food.
- You have extremely salty sweat. When you
perspire, your body loses large amounts of salt. This can upset the balance of
minerals in your blood, which can cause a heat emergency.
- The most common symptoms of CF are frequent
coughing with phlegm, frequent bouts of bronchitis and pneumonia, salty-tasting
skin, dehydration, poor growth, and infertility, mostly in men.
- The sweat test is the most common diagnostic test
for CF. It measures the amount of salt in your sweat.
- Other tests that can be used to help diagnose CF
include a chest x ray, sinus
x ray, lung function tests, analysis of
sputum cultures and/or stool samples, and genetic testing of a blood
sample.
- Prenatal genetic testing can help you find out if
your baby is likely to have CF.
- Antibiotics are the primary treatment for lung
problems in CF. They treat airway infections. Other treatments include chest
physical therapy, exercise, mucus-thinning drugs, and other medications to
reduce inflammation in your airways and help open them up.
- Lung transplantation is an option for some people
with CF.
- The digestive problems in people with CF can be
managed with nutritional therapy, enemas, mucus-thinning drugs, and medications
to reduce stomach acid.
- Ongoing medical care from a team of health care
providers who specialize in CF is important. Good self-management includes
eating a healthy diet, avoiding tobacco smoke, exercising frequently, doing
chest physical therapy every day, drinking lots of fluids, and washing your
hands often to reduce your chances of infection.
Links to Other Information About Cystic
Fibrosis
NHLBI
Other
Clinical Trials
|
