
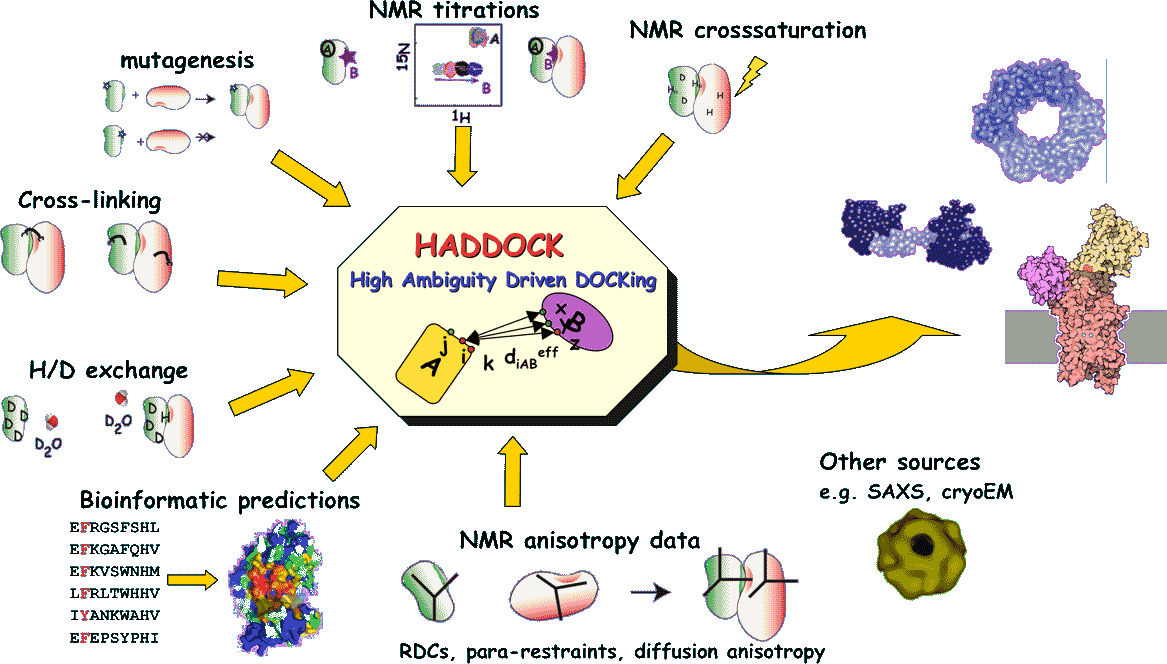
The structure determination of protein-protein complexes is a rather tedious and lengthy process, both by NMR and X-ray crystallography. Several methods based on docking to study protein complexes have been well developed over the past few years. Most of these approaches are however not driven by experimental data but based on combination of energetics and shape complementarity. HADDOCK (High Ambiguity Driven protein-protein DOCKing) is an approach that makes use of biochemical and/or biophysical interaction data such as chemical shift perturbation data resulting from NMR titration experiments or mutagenesis data.
Author: Alexandre Bonvin
How to use
Read the HADDOCK instructions carefully (particularly the Tutorial), generate the Ambiguous Interaction Restraints (AIR) file, set up a new project, and create the new.html file.
The following script generates a run.cns file from a new.html file. The run.cns file will appear in a directory run#, where # is the RUN_NUMBER from new.html.
haddock_setup.csh
#!/bin/csh # set environmental variables setenv HADDOCK /usr/local/haddock source $HADDOCK/haddock_configure.csh set runnum = `grep -P -o "^RUN_NUMBER=\d+" new.html | cut -f2 -d=` echo "run.cns is in run$runnum" haddock2.0
run.cns can be edited using a web interface. Go to the HADDOCK main page and click on 'Project setup'. Upload run.cns, edit, and save to your local disk. Replace the original run.cns.
In order to parallelize the HADDOCK run properly on Biowulf, the run.cns file that is created must be edited dynamically after nodes have been assigned (unless a multinode job is run interactively -- strongly discouraged).
The following PBS script inserts the node hostnames and the number of cpu per node into the run.cns file. Then it starts the parallelized HADDOCK run.
haddock.csh
#!/bin/csh #PBS -N haddock #PBS -o haddock.out #PBS -e haddock.err cd $PBS_O_WORKDIR # set environmental variables setenv HADDOCK /usr/local/haddock source $HADDOCK/haddock_configure.csh source /usr/local/cns/cns_solve_env set cns=$CNS_INST/bin/cns # substitute node hostnames in run.cns if (! -e run.cns-orig) then cp run.cns run.cns-orig endif set inum=1 foreach i (`cat $PBS_NODEFILE | uniq`) sed -i -e "s#queue_$inum="\""#queue_$inum="\""rsh $i csh#g" run.cns sed -i -e "s#cns_exe_$inum="\""#cns_exe_$inum="\""$cns#g" run.cns set ncpu=`rsh $i "cat /proc/cpuinfo | grep -c ^processor"` sed -i -e "s#cpunumber_$inum=0#cpunumber_$inum=$ncpu#g" run.cns @ inum+=1 end # now run parallelized HADDOCK haddock2.0 >& haddock.log
The haddock.csh PBS script is submitted from the run directory (e.g., run1) where the run.cns file was created using the qsub command
qsub -l nodes=N haddock.csh
where N is the number of nodes desired.
More information and finer details
HADDOCK is not perfectly parallelized, and is certainly not optimized for running under PBS Pro. It is most efficient to have the number of structures be a multiple of the number of CPUs desired. For example, 1000 structures in iteration 0 and 200 for iteration 1, would best be run with 20, 40, or 50 CPU. This maximizes the number of active CPU during the run, and minimizes the total time for the job to complete.
Also, there are still bugs in version 2.0. Sometimes HADDOCK will fail unexpectantly or stall at a point during the run. It is best to keep an eye on the job to make sure it doesn't go haywire.
