| | | | |
Research
|
| Inorganic Arsenite Potentiates Vasoconstriction through Calcium Sensitization in Vascular Smooth Muscle Moo-Yeol Lee,1 Young-Ho Lee,2 Kyung-Min Lim,1 Seung-Min Chung,1 Ok-Nam Bae,1 Heon Kim,3 Choong-Ryeol Lee,4 Jung-Duck Park,5 and Jin-Ho Chung1 1College of Pharmacy, Seoul National University, Seoul, Korea; 2College of Medicine and BK21 Project for Medical Sciences, Yonsei University, Seoul, Korea; 3College of Medicine, Chungbuk National University, Cheongju, Korea; 4Ulsan University Hospital, Ulsan, Korea; 5College of Medicine, Chung-Ang University, Seoul, Korea Abstract
Chronic exposure to arsenic is well known as the cause of cardiovascular diseases such as hypertension. To investigate the effect of arsenic on blood vessels, we examined whether arsenic affected the contraction of aortic rings in an isolated organ bath system. Treatment with arsenite, a trivalent inorganic species, increased vasoconstriction induced by phenylephrine or serotonin in a concentration-dependent manner. Among the arsenic species tested--arsenite, pentavalent inorganic species (arsenate) , monomethylarsonic acid (MMAV) , and dimethylarsinic acid (DMAV) --arsenite was the most potent. Similar effects were also observed in aortic rings without endothelium, suggesting that vascular smooth muscle plays a key role in enhancing vasoconstriction induced by arsenite. This hypercontraction by arsenite was well correlated with the extent of myosin light chain (MLC) phosphorylation stimulated by phenylephrine. Direct Ca2+ measurement using fura-2 dye in aortic strips revealed that arsenite enhanced vasoconstriction induced by high K+ without concomitant increase in intracellular Ca2+ elevation, suggesting that, rather than direct Ca2+ elevation, Ca2+ sensitization may be a major contributor to the enhanced vasoconstriction by arsenite. Consistent with these in vitro results, 2-hr pretreatment of 1.0 mg/kg intravenous arsenite augmented phenylephrine-induced blood pressure increase in conscious rats. All these results suggest that arsenite increases agonist-induced vasoconstriction mediated by MLC phosphorylation in smooth muscles and that calcium sensitization is one of the key mechanisms for the hypercontraction induced by arsenite in blood vessels. Key words: arsenic, arsenite, blood vessels, calcium sensitization, cardiovascular disease, myosin light chain phosphorylation, vasoconstriction. Environ Health Perspect 113:1330-1335 (2005) . doi:10.1289/ehp.8000 available via http://dx.doi.org/ [Online 14 June 2005]
Address correspondence to J.-H. Chung, College of Pharmacy, Seoul National University, Shinrim-dong San 56-1, Seoul 151-742, Korea. Telephone: 82-2-880-7856. Fax: 82-2-885-4157. E-mail: jhc302@plaza.snu.ac.kr This work was supported by the Ministry of Science and Technology National Research and Development Program and by the Eco-Technopia 21 project of the Ministry of Environment, Korea. The authors declare they have no competing financial interests. Received 7 February 2005 ; accepted 14 June 2005. |
|
|
|
Arsenic is a ubiquitous element distributed in the environment, and millions of people are chronically exposed to arsenic worldwide (Abernathy et al. 1999). Naturally contaminated drinking water is the main source of arsenic exposure, posing potential risk to human health (Nordstrom 2002; Schoen et al. 2004; Smith et al. 2002). Chronic arsenic exposure has been associated with a wide range of illnesses including cancer, hyperkeratosis, diabetes, and cardiovascular disease (Engel et al. 1994; Rossman 2003; Tseng 2004; Yu et al. 1984). Cardiovascular effects of arsenic exposure include hypertension, atherosclerosis, cerebrovascular disease, ischemic heart disease, and peripheral vascular disorders such as blackfoot disease (resulting from gangrene caused by obstruction of peripheral blood vessels) in humans (Chen et al. 1995, 1996; Chiou et al. 1997; Rahman et al. 1999; Simeonova et al. 2003; Wang et al. 2002).
Lee et al. (2002) recently suggested that the mechanism for arsenic-induced cardiovascular disease is the increased susceptibility of platelets to aggregate, resulting in enhanced arterial thrombosis. Other mechanisms may also be responsible for the diversity of human cardiovascular disease from chronic arsenic exposure. One possibility is that arsenic may alter the normal vasomotor tone of blood vessels, which rises from contractility of vascular smooth muscle cells.
The contraction of smooth muscle is regulated by mediators such as neural and humoral factors, mechanical forces, and vasoactive substances from endothelial cells. Vascular smooth muscle contraction is triggered primarily by a rise in intracellular free Ca2+ concentration (Sanders 2001). Ca2+ binds to calmodulin (CaM), allowing Ca2+-CaM complex formation, which binds to and activates myosin light chain kinase (MLCK) (Horowitz et al. 1996). The active MLCK catalyzes the phosphorylation of the regulatory myosin light chain (MLC), which then triggers myosin-actin interaction, leading to the shortening of muscle and generation of force. When the intracellular Ca2+ concentration returns to a lower level, myosin is dephosphorylated by myosin phosphatase and the muscle relaxes (Somlyo and Somlyo 1994; Walsh et al. 1994).
In addition to the contraction mediated by Ca2+-dependent MLCK, it has been recently suggested that smooth muscle contraction is modulated by Ca2+ sensitization (Somlyo and Somlyo 2000). This implies that Ca2+-dependent contractions occur, but at a lower Ca2+ concentration than would be expected for that mediated directly via MLCK (Somlyo and Somlyo 2000). An increase in MLC phosphorylation due to reduced activity of myosin phosphatase appears to result in the hypercontraction of blood vessels, which is a characteristic feature of Ca2+ sensitization (Fukata et al. 2001; Uehata et al. 1997). This abnormal hypercontraction is generally known to cause acute vasospasm, microcirculatory ischemia, increased systemic blood pressure, and ultimately, possible vascular diseases (Bohr et al. 1991; Mohri et al. 1998; Mulvany and Aalkjaer 1990).
Previous epidemiologic studies have reported that peripheral vascular resistance and systemic blood pressure were elevated in populations that had ingested the arsenic-contaminated drinking water (Chen and Yen 1964; Tseng et al. 1995). Carmignani et al. (1985) observed that chronic administration of arsenite to rats and rabbits caused a significant increase in vascular peripheral resistance. These studies suggest the possibility that arsenic may disrupt normal vasomotor function, leading to hypercontraction of blood vessels. Indeed, our previous study demonstrated that arsenic could inhibit acetylcholine-induced vascular relaxation via inhibition of nitric oxide synthase in vascular endothelial cells (Lee et al. 2003). In the process of investigating the effect of arsenic on blood vessels, we observed that arsenic could enhance agonist-induced contraction in an aortic ring organ bath system, suggesting that arsenic could disrupt contractile function in vascular smooth muscles as well. Therefore, in the present study we investigated the mechanism of arsenic-induced vascular dysfunction and its possible contribution to cardiovascular diseases.
Chemicals. The following chemicals were purchased from Sigma (St. Louis, MO, USA): sodium arsenite (trivalent inorganic arsenic), sodium arsenate (pentavalent inorganic arsenic), dimethylarsinic acid (DMAV), phenylephrine, and serotonin creatinine sulfate. We obtained monomethylarsonic acid (MMAV) from Chem Service (West Chester, PA, USA) and anti-MLC and anti-phospho-MLC antibody from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Fura-2/AM was supplied by Molecular Probe (Eugene, OR, USA), and all other reagents used were of the highest purity available.
Animals. The entire animal protocol was approved by the Ethics Committee of Animal Service Center at Seoul National University. We used male Sprague-Dawley rats (Dae Han BioLink Co., Seoul, Korea) weighing 300-400 g throughout all experiments. Before the experiments, animals were acclimated for 1 week in the laboratory animal facility maintained at constant temperature and humidity with a 12-hr light/dark cycle. Food and water were provided ad libitum.
Measurement of vasoconstriction in organ bath. Rats were sacrificed by decapitation and then exsanguinated. We carefully isolated the thoracic aorta and cut it into ring segments. Aortic rings without endothelium were prepared by gently rubbing the intimal surface of the aortic rings with a wooden stick. We then mounted the rings in four-channel organ baths filled with Krebs-Ringer (KR) solution (115.5 mM NaCl, 4.6 mM KCl, 1.2 mM KH2PO4, 1.2 mM MgSO4, 2.5 mM CaCl2, 25.0 mM NaHCO3, and 11.1 mM glucose, pH 7.4). The organ bath was continuously gassed with 95% O2/5% CO2 and maintained at 37°C. The rings were stretched gradually to an optimal resting tension of 2 g and equilibrated for 30 min. The change in tension was measured isometrically with Grass FT03 force transducers (Grass Instrument Co., Quincy, MA, USA) and recorded using the AcqKnowledge III computer program (BIOPAC Systems Inc., Goleta, CA, USA).
To investigate the effect of arsenic on vasoconstriction, we treated the aortic rings with arsenic or the vehicle (saline) in minimum essential media (MEM) with 100 U/mL penicillin and 100 µg/mL streptomycin in a 95% air/5% CO2 incubator for 14 hr at 37°C. After mounting the aortic rings pretreated with arsenic in organ baths, we induced the contraction by cumulatively adding phenylephrine or serotonin to obtain concentration-contraction curves. In the experiments of high K+-induced contraction, 100 mM KCl-containing KR solution prepared by substituting K+ with Na+ was cumulatively added to the bath to obtain the indicated final concentrations.
Measurement of MLC phosphorylation. We measured the extent of MLC phosphorylation using polyacrylamide gel electrophoresis (PAGE) and immunoblot analysis using an anti-phospho-MLC antibody, as previously described (Sakurada et al. 1998). After the aortic rings were pretreated with various concentrations of arsenite for 14 hr, 10-8 M phenylephrine was added to the organ bath for 2 min, and then ice-cold acetone with 10% trichloroacetic acid and 10 mM dithiothreitol was immediately added to stop the reaction. The aortic rings were washed with the acetone solution three times, lyophilized overnight, and stored at -70°C until protein extraction. After the dried tissues were cut into small pieces, we extracted proteins in a 50 µL sample buffer containing 8 M urea, 2% sodium dodecyl sulfate, 5% β -mercaptoethanol, 0.01% bromophenol blue, and 62.5 mM Tris-HCl by vortexing for 3 hr at room temperature. Protein extracts were electrophoresed in a 15% polyacrylamide mini-slab gel (Bio-Rad, Hercules, CA, USA) and then transferred onto nitrocellulose membranes in Tris/glycine buffer (25 mM Tris, 192 mM glycine). We detected the MLC level and the extent of phosphorylation with immunoblotting using anti-MLC antibody and anti-phospho-MLC antibody (Santa Cruz Biotechnology), respectively. Immunoreactive bands were visualized by horseradish peroxidase-conjugated secondary antibody (Santa Cruz Biotechnology) and an enhanced chemiluminescence kit (ECL; Amersham, Buckinghamshire, UK). For densitometric analysis of MLC-P/MLC, we determined densities of the corresponding bands using TINA software (Raytest, Straubenhardt, Germany).
Measurement of intracellular calcium level. To measure intracellular Ca2+ levels, we cut the thoracic aorta without endothelium into spiral strips approximately 8 mm in length and 1 mm in width under a dissecting microscope. After the aortic strips were treated with arsenite for 14 hr, they were exposed to 10 µM acetoxymethyl ester of fura-2 (fura-2/AM) and 0.1% cremophor EL in a Krebs-Henseleit solution [(KH) solution: 119 mM NaCl, 4.6 mM KCl, 1.2 mM KH2PO4, 1.5 mM MgSO4, 2.5 mM CaCl2, 25.0 mM NaHCO3, and 11 mM glucose, pH 7.4] for 4 hr at room temperature.
We measured the intracellular free Ca2+ level based on the method described by Ozaki et al. (1991). Fure-2-loaded muscle strips were held horizontally in the organ chamber of a fluorimeter (CAF-110; Jasco, Tokyo, Japan) filled with KH solution. One end of the muscle strip was connected to a force-displacement transducer to monitor vessel tones. The KH solution was maintained at 37°C and continuously aerated with 95% O2/5% CO2. Passive tension of 2 g was applied and allowed to equilibrate before measurement. We elicited muscle contractions by changing the media with the KH solution containing 12.5, 25, and 90 mM KCl. Muscle strips were illuminated alternatively at 48 Hz in excitation wavelengths of 340 and 380 nm. We measured the intensity of 500 nm fluorescence emitted by 340 nm excitation (F340) and that emitted by 380 nm (F380) successively. We calculated the ratio of F340 to F380 [R(F340/F380)] as an indicator of intracellular Ca2+.
Measurement of blood pressure change induced by phenylephrine. Rats were anesthetized with phenobarbital (50 mg/kg, intraperitoneal). We placed a catheter of polyethylene PE-50 tubing (Clay Adams, Sparks, MD, USA) filled with heparinized saline (100 U/mL) in the carotid artery to measure blood pressure, and we placed a catheter of polyethylene PE-10 fused to PE-50 tubing in the jugular vein to administer drugs. Catheters were tunneled subcutaneously and exteriorized at the back of the neck. Wounds were sutured and cleaned with alcohol. Experiments were performed after a 1-day recovery period. On the day of the experiment, the arterial catheter was connected to a pressure transducer (BIOPAC Systems Inc.) and blood pressure was measured using the AcqKnowledge III computer program. We allowed blood pressure to stabilize for a minimum of 30 min before beginning treatment. To determine the effects of arsenite on blood pressure increase induced by phenylephrine, we administered arsenite solution (1.0 mg/kg) by an intravenous bolus injection into the jugular vein. In the controls, equivalent amounts of saline were injected. After 2 hr, we infused the rats with 2.5 µg/kg/min phenylephrine for 3 min via the jugular vein and monitored the change of blood pressure in response to phenylephrine simultaneously. Infusions were performed with a Harvard syringe pump (South Natick, MA, USA) at a rate of 0.1 mL/min.
Statistical analysis. We calculated the means and standard errors for all treatment groups. The data were subjected to one- or two-way analysis of variance (ANOVA) followed by Duncan's multiple range test to determine which means were significantly different from the control. Statistical analysis was performed using SPSS software (SPSS Inc., Chicago, Il, USA). In all cases, we used a p-value of < 0.05 to determine significance.
|

Figure 1. Enhancement of phenylephrine- and serotonin-induced vasoconstriction in aortic rings by arsenite. (A) Phenylephrine. (B) Serotonin. See "Materials and Methods" for details. Values shown are mean ± SE of four independent experiments.
*Significantly different from the corresponding control (p < 0.05).
|
|
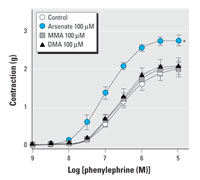
Figure 2. Effect of arsenic species on contraction of aortic rings induced by phenylephrine. See "Materials and Methods" for details. Values shown are mean ± SE of five independent experiments.
*Significantly different from the control (p < 0.05).
|
|
Figure 3. Enhancement of phenylephrine- and serotonin-induced contraction by arsenite in aortic rings without endothelium. (A) Phenylephrine (3 x 10-8 M). (B) Serotonin (10-6 M). See "Materials and Methods" for details. Values shown are mean ± SE of four independent experiments.
*Significantly different from the control (p < 0.05).
|
|

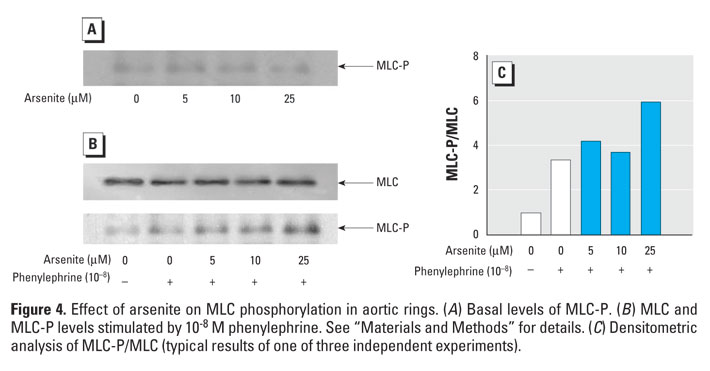
Figure 4. Effect of arsenite on MLC phosphorylation in aortic rings. (A) Basal levels of MLC-P. (B) MLC and MLC-P levels stimulated by 10-8 M phenylephrine. See "Materials and Methods" for details. (C) Densitometric analysis of MLC-P/MLC (typical results of one of three independent experiments).
|
|
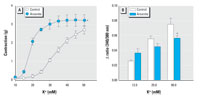
Figure 5. Effect of arsenite on vasoconstriction and intracellular Ca2+ levels in the presence of high K+. (A) Contraction induced by various concentrations of K+ in Ca2+-free KR solution in aortic rings without endothelium after treatment with arsenite. (B) Intracellular Ca2+ levels determined in the presence of K+ in fura-2 loaded aortic strips without endothelium after treatment with arsenite; values shown are mean ± SE of more than three independent experiments. See "Materials and Methods" for details.
*Significantly different from the control group (p < 0.05).
|
|
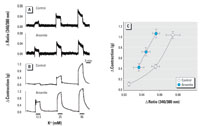
Figure 6. Simultaneous measurement of Ca2+ increase and contraction by KCl in aortic strips without endothelium after treatment with arsenite. Representative tracings of intracellular Ca2+ increase (A) and contraction (B) induced by KCl. See "Materials and Methods" for details. (C) Ca2+ elevation versus the magnitude of contraction; values shown are mean ± SE of seven independent experiments.
|
|

Figure 7. Effect of intravenously administered arsenite on phenylephrine-induced blood pressure increase in rats. (A) Saline 2 hr after arsenite exposure. (B) Phenylephrine 2 hr after saline exposure ( mmHg = 23.0 ± 3.2; mean ± SE of four animals). (C) Phenylephrine 2 hr after arsenite exposure (mmHg = 36.8 ± 3.6; mean ± SE of four animals). See "Materials and Methods" for details. Periods of infusion are indicated in each panel. Data are representative tracings of four independent experiments. mmHg = 23.0 ± 3.2; mean ± SE of four animals). (C) Phenylephrine 2 hr after arsenite exposure (mmHg = 36.8 ± 3.6; mean ± SE of four animals). See "Materials and Methods" for details. Periods of infusion are indicated in each panel. Data are representative tracings of four independent experiments.
|
|
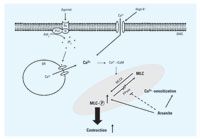
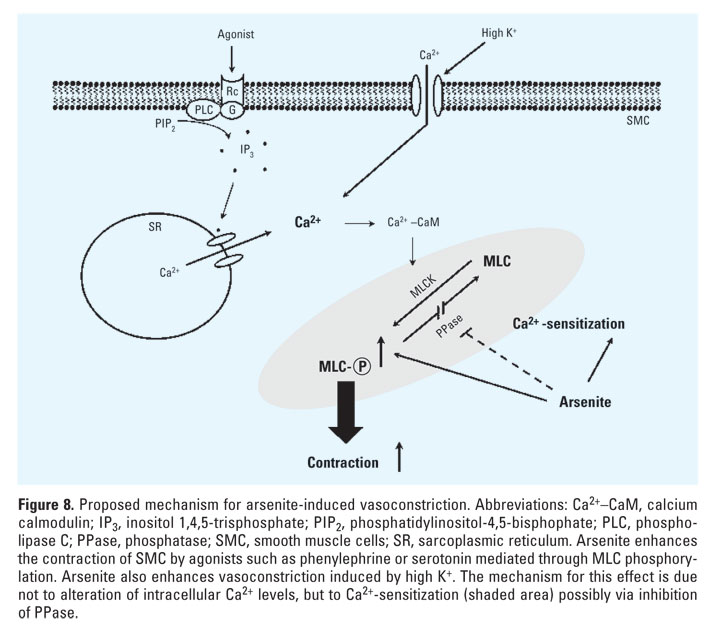
Figure 8. Proposed mechanism for arsenite-induced vasoconstriction. Abbreviations: Ca2+-CaM, calcium calmodulin; IP3, inositol 1,4,5-trisphosphate; PIP2, phosphatidylinositol-4,5-bisphophate; PLC, phospholipase C; PPase, phosphatase; SMC, smooth muscle cells; SR, sarcoplasmic reticulum. Arsenite enhances the contraction of SMC by agonists such as phenylephrine or serotonin mediated through MLC phosphorylation. Arsenite also enhances vasoconstriction induced by high K+. The mechanism for this effect is due not to alteration of intracellular Ca2+ levels, but to Ca2+-sensitization (shaded area) possibly via inhibition of PPase.
|
To investigate whether arsenic affects contraction of blood vessels, we treated intact aortic rings with various concentrations of arsenite (trivalent inorganic arsenic) for 14 hr and then added phenylephrine and serotonin cumulatively to obtain concentration-contraction curves. Arsenite alone did not cause any changes in basal vascular tone (data not shown). Arsenite treatment, however, enhanced the contraction induced by both phenylephrine and serotonin in a concentration-dependent manner (Figure 1). We investigated the effects of pentavalent inorganic species (arsenate) and two major metabolites, MMAV and DMAV, on phenylephrine-induced constriction (Figure 2). Arsenate did enhance phenylephrine-induced vasoconstriction, but the concentration required was higher than that of arsenite. MMAV and DMAV up to 100 µM failed to affect the phenylephrine-induced vasoconstriction. These results suggest that agonist-induced contraction in blood vessels can be enhanced by arsenic and that arsenite is the most potent form tested.
To investigate whether the enhanced contraction induced by arsenite was an endothelium-dependent effect, we performed experiments using aortic rings without endothelium. Treatment with arsenite to aortic rings without endothelium still resulted in a concentration-dependent increment of agonist-induced contraction in blood vessels (Figure 3), suggesting that the enhanced contraction by arsenite is primarily due to hypercontraction of smooth muscles. To examine whether hypercontraction by arsenite is mediated by the phosphorylation of MLC in smooth muscles, we evaluated the effect of arsenite on phenylephrine-induced MLC phosphorylation in aortic rings without endothelium. Arsenite treatment alone did not alter the basal levels of MLC phosphorylation (Figure 4A). Addition of phenylephrine alone did not affect the total MLC levels, but it increased MLC phosphorylation significantly (Figure 4B,C). However, when the aortic rings without endothelium were stimulated with phenylephrine, arsenite treatment resulted in a significant increase in MLC phosphorylation without a concomitant change in MLC levels (Figure 4B,C). These results indicate that arsenite enhances agonist-induced vasoconstriction through MLC phosphorylation in smooth muscles.
To examine whether the increased MLC phosphorylation was mediated by intracellular Ca2+ elevation in vascular smooth muscles, we investigated the effects of arsenic on intracellular Ca2+ levels when vasoconstriction was initiated by high K+ concentration. We used a high concentration of K+ to induce contraction in blood vessels, since K+ directly induces rapid influx of extracellular Ca2+ through voltage-gated Ca2+ channels in plasma membrane without involving other signal transduction pathway and thus could serve as a simple alternative tool to investigate the current premise. As shown in Figure 5A, 25 µM arsenite enhanced vasoconstriction induced by 10-50 mM K+ in aortic rings without endothelium, which is consistent with the results shown in Figure 3. The next experiment was performed using aortic strips loaded with fura-2 fluorescent dye to investigate whether arsenic increased intracellular Ca2+ in the presence of high K+. Arsenite, however, did not induce intracellular Ca2+ elevation, but rather decreased Ca2+ concentration significantly in the presence of 90 mM K+ (Figure 5B). These results show that arsenite enhances vasoconstriction without concomitant increase of intracellular Ca2+ levels, suggesting that arsenite might increase the contractility of blood vessels via Ca2+ sensitization.
To investigate this assumption, we measured smooth muscle contraction and the change of intracellular Ca2+ levels simultaneously using fura-2-loaded aortic strips. After the muscle strips were treated with 25 µM arsenite, contraction was induced successively by 12.5, 25, and 90 mM K+. Figure 6A and 6B shows a scanned reduction of the tracings from aortic strips. Compared with the control group, arsenite-treated aortic strips showed enhanced contraction without any concomitant increase in intracellular Ca2+ elevation (Figure 6A,B). Plotting this result reveals that the aortic strips treated with arsenite showed a steeper slope in the intracellular Ca2+ elevation versus contraction relationship (Figure 6C). These results suggest that arsenite might potentiate vasoconstriction by enhancing the Ca2+ sensitivity of contractile machinery in smooth muscle.
To verify the effects of arsenite on blood vessels invivo, we monitored changes in blood pressure after intravenous infusion of phenylephrine into conscious rats (Figure 7). An intravenous bolus of arsenite had no effect on basal blood pressure. When rats were treated with arsenite 2 hr before phenylephrine infusion, the hypertensive effect of phenylephrine was significantly potentiated (23.0 ± 3.2 vs. 36.8 ± 3.6 mmHg, p = 0.029; Figure 7B,C). These results suggest that arsenite could induce the enhancement of agonist-induced vasoconstriction invivo and this confirms the previous invitro results (Figure 1).
In the present study we demonstrate the ability of arsenic to enhance contraction of isolated aortic rings from rats. We have shown that arsenite enhances the vascular contraction induced by phenylephrine, serotonin, and high K+ in a concentration-dependent manner and that the Ca2+ sensitization in smooth muscle largely contributes to arsenite-induced hypercontractility. These in vitro results were consistent with in vivo results in which arsenite potentiated the hypertensive effect of phenylephrine. Recently, the effect of arsenic on platelets has been suggested as a key mechanism in the development of cardiovascular diseases (Lee et al. 2002). Blood vessels, however, are another important tissue in cardiovascular system. Hypercontractility of blood vessels disrupts vasomotor tone regulation and makes vasoconstriction predominate, which can induce hypertension, complicating cardiovascular disease. Elevated peripheral resistance has been reported in populations exposed to arsenic-contaminated drinking water and in animals treated with arsenite (Carmignani et al. 1985; Chen and Yen 1964). Our data provide evidence that arsenite could enhance vascular smooth muscle contractility, suggesting that arsenic-induced hypercontraction of blood vessels might be another mechanism for arsenic-associated cardiovascular disease observed in human populations.
Suppression of nitric oxide production in endothelium results in the loss of vasodilator activity, causing vasoconstriction (Moncada et al. 1991), and we previously demonstrated that arsenite disrupts endothelium-dependent vasorelaxation via inhibition of endothelial nitric oxide production (Lee et al. 2002). Pi et al. (2003) also reported that vasoconstriction was increased in aortic rings isolated from arsenate-exposed rabbits. They explained the phenomena as a result of impaired nitric oxide formation. To examine whether the hypercontraction of blood vessels observed (Figure 1) was also mediated by the inhibition of endothelium-derived vasorelaxation activity, we performed experiments in aortic rings without endothelium. Surprisingly, contraction by phenylephrine or serotonin was still enhanced by arsenite treatment (Figure 3), suggesting that the hypercontraction was dependent on smooth muscles in blood vessels. Because arsenite not only interferes with endothelium-dependent vasorelaxation but also enhances smooth muscle-dependent contraction, arsenite treatment could result in overall hypercontraction of blood vessels, leading to a possible increased risk for development of vascular diseases such as hypertension and atherosclerosis (Bohr et al. 1991; Mulvany and Aalkjaer 1990; Rubanyi 1993; Vanhoutte 1997).
As shown in Figure 2, treatment with arsenate (pentavalent inorganic arsenic)also potentiated phenylephrine-induced contraction, whereas MMAV and DMAV showed no significant effect. Arsenate is generally known to exert its toxic effects by replacing phosphate in various biochemical reactions because it has a similar structure and properties to phosphate (Oremland and Stolz 2003). However, it is not certain that such properties are engaged in the enhancement of vasoconstriction shown in this study. Indeed, arsenate can be reduced to arsenite at a comparable rate in cell systems, and the biological effect of arsenate may in part result from its reduction to arsenite (Huang and Lee 1996). Therefore, considering that the effective concentration of arsenate is higher than that of arsenite, it is possible that the effect of arsenate arises from arsenite formed by reduction of arsenate.
Phenylephrine and serotonin act on the  1-adrenoceptor and the 5-HT2 serotonin receptor, respectively, on aortic smooth muscle cells and thus result in increasing intracellular Ca2+ (Bruckner et al. 1984; Peroutka 1984). In contrast to these receptor agonists, a high concentration of K+ bypasses receptor-signaling pathways and leads directly to intracellular Ca2+ elevation by opening Ca2+ channels following membrane depolarization (Chiu et al. 1987). These intracellular Ca2+ increases by phenylephrine, serotonin, and high K+ result in phosphorylation of MLC leading to vascular smooth muscle contraction. It is widely accepted that the degree of MLC phosphorylation is the essential factor that determines the extent to which smooth muscle contracts (Fukata et al. 2001; Walsh 1994). As shown in Figure 4B and 4C, MLC phosphorylation to phenylephrine was augmented by arsenite, from which it can be concluded that hypercontraction by arsenite is an MLC phosphorylation-dependent effect. In contrast, arsenite alone did not induce the MLC phosphorylation significantly (Figure 4A), consistent with the fact that basal tones of blood vessels were not changed by arsenite treatment alone. 1-adrenoceptor and the 5-HT2 serotonin receptor, respectively, on aortic smooth muscle cells and thus result in increasing intracellular Ca2+ (Bruckner et al. 1984; Peroutka 1984). In contrast to these receptor agonists, a high concentration of K+ bypasses receptor-signaling pathways and leads directly to intracellular Ca2+ elevation by opening Ca2+ channels following membrane depolarization (Chiu et al. 1987). These intracellular Ca2+ increases by phenylephrine, serotonin, and high K+ result in phosphorylation of MLC leading to vascular smooth muscle contraction. It is widely accepted that the degree of MLC phosphorylation is the essential factor that determines the extent to which smooth muscle contracts (Fukata et al. 2001; Walsh 1994). As shown in Figure 4B and 4C, MLC phosphorylation to phenylephrine was augmented by arsenite, from which it can be concluded that hypercontraction by arsenite is an MLC phosphorylation-dependent effect. In contrast, arsenite alone did not induce the MLC phosphorylation significantly (Figure 4A), consistent with the fact that basal tones of blood vessels were not changed by arsenite treatment alone.
In addition to intracellular Ca2+ elevation, the level of responsiveness of contractile machinery to intracellular Ca2+ plays a key role in regulating the contractility of smooth muscle cells (Somlyo and Somlyo 2000). Simultaneous measurement of contraction and intracellular Ca2+ increase in aortic strips revealed that arsenite potentiated the magnitude of contraction without concomitant increase in Ca2+ elevation (Figure 6), suggesting significant contribution of Ca2+ sensitization to the hypercontraction induced by arsenite. Previous studies suggested that the mechanism for Ca2+ sensitization was mainly due to modulation of MLC phosphatase activity (Fukata et al. 2001; Uehata et al. 1997). That is, if MLC phosphatase was inhibited, the enhanced MLC phosphorylation could be elicited at the same Ca2+ concentration, which resulted in hypercontraction. However, it is currently uncertain whether arsenite could inhibit MLC phosphatase directly.
In this study, we showed that arsenite causes enhanced vasoconstriction in vitro and demonstrated that Ca2+ sensitization in smooth muscle is responsible for MLC phosphorylation-dependent hypercontraction by arsenite (Figure 8). In our in vivo study, arsenite treatment potentiated the hypertensive effect of phenylephrine in rats. These results confirm our in vitro observations and suggest that increased vasocontraction may be a contributing factor in the development of cardiovascular diseases in populations exposed to arsenic.
|
|
|
| [References Listed in PubMed] References
Abernathy CO, Liu YP, Longfellow D, Aposhian HV, Beck B, Fowler B, et al. 1999. Arsenic health effects, mechanisms of actions, and research issues. Environ Health Perspect 107:593-597.
Bohr DF, Dominiczak AF, Webb RC. 1991. Pathophysiology of the vasculature in hypertension. Hypertension 18(5 suppl):III69-75.
Bruckner R, Meyer W, Mugge A, Schmitz W, Scholz H. 1984. Alpha-adrenoceptor-mediated positive inotropic effect of phenylephrine in isolated human ventricular myocardium. Eur J Pharmacol 99:345-347.
Carmignani M, Boscolo P, Castellino N. 1985. Metabolic fate and cardiovascular effects of arsenic in rats and rabbits chronically exposed to trivalent and pentavalent arsenic. Arch Toxicol Suppl 8:452-455.
Chen CJ, Chiou HY, Chiang MH, Lin LJ, Tai TY. 1996. Dose-response relationship between ischemic heart disease mortality and long-term arsenic exposure. Arterioscler Thromb Vasc Biol 16:504-510.
Chen CJ, Hsueh YM, Lai MS, Shyu MP, Chen SY, Wu MM, et al. 1995. Increased prevalence of hypertension and long-term arsenic exposure. Hypertension 25:53-60.
Chen WY, Yen TS. 1964. Experimental studies on the drinking water of blackfoot endemic area. 2. Studies on the effects of drinking water of blackfoot endemic area on peripheral vascular perfusion of the hind limbs of frogs. Tsa Chih Gaoxiong Yi Xue Yuan Tong Xue Hui 63:150-158.
Chiou HY, Huang WI, Su CL, Chang SF, Hsu YH, Chen CJ. 1997. Dose-response relationship between prevalence of cerebrovascular disease and ingested inorganic arsenic. Stroke 28:1717-1723.
Chiu AT, Bozarth JM, Timmermans PB. 1987. Relationship between phosphatidylinositol turnover and Ca++ mobilization induced by alpha-1 adrenoceptor stimulation in the rat aorta. J Pharmacol Exp Ther 240:123-127.
Engel RR, Hopenhayn-Rich C, Receveur O, Smith AH. 1994. Vascular effects of chronic arsenic exposure: a review. Epidemiol Rev 16:184-209.
Fukata Y, Amano M, Kaibuchi K. 2001. Rho-Rho-kinase pathway in smooth muscle contraction and cytoskeletal reorganization of non-muscle cells. Trends Pharmacol Sci 22:32-39.
Horowitz A, Menice CB, Laporte R, Morgan KG. 1996. Mechanisms of smooth muscle contraction. Physiol Rev 76:967-1003.
Huang RN, Lee TC. 1996. Cellular uptake of trivalent arsenite and pentavalent arsenate in KB cells cultured in phosphate-free medium. Toxicol Appl Pharmacol 136:243-249.
Lee MY, Bae ON, Chung SM, Kang KT, Lee JY, Chung JH. 2002. Enhancement of platelet aggregation and thrombus formation by arsenic in drinking water: a contributing factor to cardiovascular disease. Toxicol Appl Pharmacol 179:83-88.
Lee MY, Jung BI, Chung SM, Bae ON, Lee JY, Park JD, et al. 2003. Arsenic-induced dysfunction in relaxation of blood vessels. Environ Health Perspect 111:513-517.
Mohri M, Koyanagi M, Egashira K, Tagawa H, Ichiki T, Shimokawa H, et al. 1998. Angina pectoris caused by coronary microvascular spasm. Lancet 351:1165-1169.
Moncada S, Rees DD, Schulz R, Palmer RM. 1991. Development and mechanism of a specific supersensitivity to nitrovasodilators after inhibition of vascular nitric oxide synthesis in vivo. Proc Natl Acad Sci USA 88:2166-2170.
Mulvany MJ, Aalkjaer C. 1990. Structure and function of small arteries. Physiol Rev 70:921-961.
Nordstrom DK. 2002. Public health. Worldwide occurrences of arsenic in ground water. Science 296:2143-2145. [CrossRef].
Oremland RS, Stolz JF. 2003. The ecology of arsenic. Science 300:939-944.
Ozaki H, Stevens RJ, Blondfield DP, Publicover NG, Sanders KM. 1991. Simultaneous measurement of membrane potential, cytosolic Ca2+, and tension in intact smooth muscles. Am J Physiol 260:C917-C925.
Peroutka SJ. 1984. Vascular serotonin receptors. Correlation with 5-HT1 and 5-HT2 binding sites. Biochem Pharmacol 33:2349-2353. [CrossRef].
Pi J, Horiguchi S, Sun Y, Nikaido M, Shimojo N, Hayashi T, et al. 2003. A potential mechanism for the impairment of nitric oxide formation caused by prolonged oral exposure to arsenate in rabbits. Free Radic Biol Med 35:102-113.
Rahman M, Tondel M, Ahmad SA, Chowdhury IA, Faruquee MH, Axelson O. 1999. Hypertension and arsenic exposure in Bangladesh. Hypertension 33:74-78.
Rossman TG. 2003. Mechanism of arsenic carcinogenesis: an integrated approach. Mutat Res 533:37-65.
Rubanyi GM. 1993. The role of endothelium in cardiovascular homeostasis and diseases. J Cardiovasc Pharmacol 22(suppl 4):S1-S14.
Sakurada K, Seto M, Sasaki Y. 1998. Dynamics of myosin light chain phosphorylation at Ser19 and Thr18/Ser19 in smooth muscle cells in culture. Am J Physiol 274:C1563-C1572.
Sanders KM. 2001. Invited review: mechanisms of calcium handling in smooth muscles. J Appl Physiol 91:1438-1449.
Schoen A, Beck B, Sharma R, Dube E. 2004. Arsenic toxicity at low doses: epidemiological and mode of action considerations. Toxicol Appl Pharmacol 198:253-267.
Simeonova PP, Hulderman T, Harki D, Luster MI. 2003. Arsenic exposure accelerates atherogenesis in apolipoprotein E(-/-) mice. Environ Health Perspect 111:1744-1748.
Smith AH, Lopipero PA, Bates MN, Steinmaus CM. 2002. Public health. Arsenic epidemiology and drinking water standards. Science 296:2145-2146.
Somlyo AP, Somlyo AV. 1994. Signal transduction and regulation in smooth muscle. Nature 372:231-236. [CrossRef].
Somlyo AP, Somlyo AV. 2000. Signal transduction by G-proteins, rho-kinase and protein phosphatase to smooth muscle and non-muscle myosin II. J Physiol 522(Pt 2):177-185. [CrossRef].
Tseng CH. 2004. The potential biological mechanisms of arsenic-induced diabetes mellitus. Toxicol Appl Pharmacol 197:67-83. [CrossRef].
Tseng CH, Chong CK, Chen CJ, Lin BJ, Tai TY. 1995. Abnormal peripheral microcirculation in seemingly normal subjects living in blackfoot-disease-hyperendemic villages in Taiwan. Int J Microcirc Clin Exp 15:21-27.
Tseng WP. 1977. Effects and dose-response relationships of skin cancer and blackfoot disease with arsenic. Environ Health Perspect 19:109-119.
Uehata M, Ishizaki T, Satoh H, Ono T, Kawahara T, Morishita T, et al. 1997. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature 389:990-994.
Vanhoutte PM. 1997. Endothelial dysfunction and atherosclerosis. Eur Heart J 18(suppl E):E19-E29.
Walsh MP. 1994. Regulation of vascular smooth muscle tone. Can J Physiol Pharmacol 72:919-936.
Wang CH, Jeng JS, Yip PK, Chen CL, Hsu LI, Hsueh YM, et al. 2002. Biological gradient between long-term arsenic exposure and carotid atherosclerosis. Circulation 105:1804-1809.
Yu HS, Sheu HM, Ko SS, Chiang LC, Chien CH, Lin SM, et al. 1984. Studies on blackfoot disease and chronic arsenism in southern Taiwan: with special reference to skin lesions and fluorescent substances. J Dermatol 11:361-370.
Last Updated: April 25, 2006
|
|
|
|
| |