
| | | | |
Research
|
Lead Increases Lipopolysaccharide-Induced Liver-Injury Through Tumor Necrosis Factor-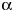 Overexpression by Monocytes/Macrophages: Role of Protein Kinase C and P42/44 Mitogen-Activated Protein Kinase Overexpression by Monocytes/Macrophages: Role of Protein Kinase C and P42/44 Mitogen-Activated Protein Kinase Yu-Jung Cheng,1,2 Bei-Chang Yang,2,3 and Ming-Yie
Liu1,2 1Department of Environmental and Occupational Health, 2Institute
of Basic Medical Sciences, and 3Department of Microbiology and Immunology,
National Cheng Kung University Medical College, Tainan, Taiwan Abstract
Although lead and lipopolysaccharide (LPS) , both important environmental pollutants, activate cells through different receptors and participate in distinct upstream signaling pathways, Pb increases the amount of LPS-induced tumor necrosis factor-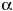 (TNF-) . We examined the cells responsible for the excess production of Pb-increased LPS-induced TNF- and liver injury, and the roles of protein kinase C (PKC) and p42/44 mitogen-activated protein kinase (MAPK) in the induction of TNF-. Peritoneal injection of Pb alone (100 µmol/kg) or a low dose of LPS (5 mg/kg) did not affect serum TNF- or liver functions in A/J mice. In contrast, coexposure to these noneffective doses of Pb plus LPS (Pb+LPS) strongly induced TNF- expression and resulted in profound liver injury. Direct inhibition of TNF- or functional inactivation of monocytes/macrophages significantly decreased the level of Pb+LPS-induced serum TNF- and concurrently ameliorated liver injury. Pb+LPS coexposure stimulated the phosphorylation of p42/44 MAPK and the expression of TNF- in CD14+ cells of cultured mouse whole blood, peritoneal macrophages, and RAW264.7 cells. Moreover, blocking PKC or MAPK effectively reduced Pb+LPS-induced TNF- expression and liver injury. In summary, monocytes/macrophages were the cells primarily responsible for producing, through the PKC/MAPK pathway, the excess Pb-increased/LPS-induced TNF- that caused liver injury. Key words: lead, lipopolysaccharide, liver injury, monocytes/macrophage, p42/44 mitogen-activated protein kinase, protein kinase C, tumor necrosis factor-. Environ Health Perspect 114: 507-513 (2006) . doi:10.1289/ehp.8550 available via http://dx.doi.org/ [Online 10 November 2005] (TNF-) . We examined the cells responsible for the excess production of Pb-increased LPS-induced TNF- and liver injury, and the roles of protein kinase C (PKC) and p42/44 mitogen-activated protein kinase (MAPK) in the induction of TNF-. Peritoneal injection of Pb alone (100 µmol/kg) or a low dose of LPS (5 mg/kg) did not affect serum TNF- or liver functions in A/J mice. In contrast, coexposure to these noneffective doses of Pb plus LPS (Pb+LPS) strongly induced TNF- expression and resulted in profound liver injury. Direct inhibition of TNF- or functional inactivation of monocytes/macrophages significantly decreased the level of Pb+LPS-induced serum TNF- and concurrently ameliorated liver injury. Pb+LPS coexposure stimulated the phosphorylation of p42/44 MAPK and the expression of TNF- in CD14+ cells of cultured mouse whole blood, peritoneal macrophages, and RAW264.7 cells. Moreover, blocking PKC or MAPK effectively reduced Pb+LPS-induced TNF- expression and liver injury. In summary, monocytes/macrophages were the cells primarily responsible for producing, through the PKC/MAPK pathway, the excess Pb-increased/LPS-induced TNF- that caused liver injury. Key words: lead, lipopolysaccharide, liver injury, monocytes/macrophage, p42/44 mitogen-activated protein kinase, protein kinase C, tumor necrosis factor-. Environ Health Perspect 114: 507-513 (2006) . doi:10.1289/ehp.8550 available via http://dx.doi.org/ [Online 10 November 2005]
Address correspondence to B.-C. Yang, Department of Microbiology and Immunology, National Cheng Kung University Medical College, Tainan 70428, Taiwan. Telephone: 886-6-235-3535 Ext. 5637. Fax: 886-6-208-2705. E-mail: y1357@mail.ncku.edu.tw We thank S.-T. Wang for comments on statistical analysis and B. Franke for editorial assistance. This study was supported in part by grant NSC-94-2211-E-006-090 to M.-Y.L. (co-correspondence) and grant NSC-94-2314-B-006-050 to B.-C.Y. from the National Science Council, Taiwan. The authors declare they have no competing financial interests. Received 30 July 2005 ; accepted 9 November 2005. |
|
|
|
Tumor necrosis factor- (TNF- )
regulates a variety of biologic functions,
including organ development, immune homeostasis,
and malignance. The body subtly regulates
the expression kinetics and dose of TNF- to
ensure its proper effect because TNF- has
opposite biologic effects in different circumstances
(Aggarwal 2003; Pfeffer 2003). On the one
hand, TNF- is
essential for the host in tissue repair and
in protective immune responses against infection.
On the other hand, inadequate TNF- may
have detrimental consequences in sepsis,
tumor formation, and autoimmune diseases.
Regulating the expression of TNF- has
been an important subject in managing acute
inflammatory diseases that include bacterial
sepsis (Spooner et al. 1992). Recent studies
on chronic neuronal disease revealed a new
feature of inflammation: a transient spike
of TNF- expression
(i.e., a large amount that peaks after 1.5
hr and disappears after 3 hr) may induce
neuronal degeneration resembling the delayed
and progressive nature of the symptoms in
patients with Parkinson’s disease (Gao
et al. 2002). These findings indicate that
the extent of TNF- expression
during a disease determines not only severity
and survival rate but also delayed disease
sequelae. The in vivo lipopolysaccharide (LPS)-induced
model of liver damage of mice, used to determine
TNF--mediated
organ failure, is both sensitive and convenient.
LPS, a component of the outer membrane of
gram-negative bacteria, plays a major role
in inducing septic shock and is a potent
inducer of TNF-in
vivo and in vitro (Goldfeld et
al. 1990; Ulich et al. 1991). The binding
of LPS to CD14/LPS-binding protein and Toll-like
receptor-4 triggers multiple signal cascades
that activate nuclear factor-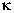 B
and p42/44 mitogen-activated protein kinase
(MAPK) and lead to the expression of proinflammatory
cytokines, including TNF- (Triantafilou
and Triantafilou 2002). LPS causes liver
injury at high doses (Kosai et al. 1999)
but a modest, noninjurious inflammation at
low doses (Ganey and Roth 2001) in several
animal models. High-dose LPS-induced liver
injury is partly attributed to excess TNF- production
(Hewett et al. 1993). TNF--associated
signal transduction has been well characterized.
TNF- might
trigger apoptosis in hepatocytes by signaling
through the Fas-associated death-domain protein
of the TNF receptor that activates caspases
(Leist et al. 1996; Schuchmann et al. 2003).
Blocking TNF production or trimming the signaling
pathway using caspase-inhibitors reduces
TNF--mediated
liver injury (Kunstle et al. 1997). In addition,
LPS induces apoptosis in macrophages through
TNF- (Comalada
et al. 2003). Convincing evidence shows that
metal pollutants in the living environment
may modulate the effects of LPS. Among them,
lead, an important industrial pollutant,
not only altered the immune response (Luster
et al. 1978) but also increased the mortality
of an LPS challenge or bacterial infections
in several animal studies (Dentener et al.
1989; Selye et al. 1966). Thus, the influence
of environmental factors on TNF- production
is a significant issue. B
and p42/44 mitogen-activated protein kinase
(MAPK) and lead to the expression of proinflammatory
cytokines, including TNF- (Triantafilou
and Triantafilou 2002). LPS causes liver
injury at high doses (Kosai et al. 1999)
but a modest, noninjurious inflammation at
low doses (Ganey and Roth 2001) in several
animal models. High-dose LPS-induced liver
injury is partly attributed to excess TNF- production
(Hewett et al. 1993). TNF--associated
signal transduction has been well characterized.
TNF- might
trigger apoptosis in hepatocytes by signaling
through the Fas-associated death-domain protein
of the TNF receptor that activates caspases
(Leist et al. 1996; Schuchmann et al. 2003).
Blocking TNF production or trimming the signaling
pathway using caspase-inhibitors reduces
TNF--mediated
liver injury (Kunstle et al. 1997). In addition,
LPS induces apoptosis in macrophages through
TNF- (Comalada
et al. 2003). Convincing evidence shows that
metal pollutants in the living environment
may modulate the effects of LPS. Among them,
lead, an important industrial pollutant,
not only altered the immune response (Luster
et al. 1978) but also increased the mortality
of an LPS challenge or bacterial infections
in several animal studies (Dentener et al.
1989; Selye et al. 1966). Thus, the influence
of environmental factors on TNF- production
is a significant issue.
Several pieces of evidence (Liu et al.
2001) suggest that Pb might act on calcium
channels to alter intracellular calcium homeostasis
in bone cells (Pounds 1984; Rosen and Pounds
1989; Schanne et al. 1989) and neuronal cells
(Goldstein 1993; Pounds 1984; Rosen and Pounds
1989; Schanne et al. 1989). Although the
cellular target of Pb is still elusive, exposure
to Pb activates protein kinase C (PKC) in
several types of cells, such as astrocytes
and neuronal cells in the brain (Costa 1998;
Markovac and Goldstein 1988). We previously
(Cheng et al. 2004) demonstrated that Pb
stimulates PKC to activate p42/44 MAPK, which
results in the expression of TNF- in
glial cells. Although LPS and Pb trigger
intracellular signals by different routes,
Pb increases LPS-induced TNF- production
(Liu et al. 2005). Coexposure to Pb plus
LPS also induces TNF- expression
through PKC and p42/44 MAPK, causing liver
injury in rats (Cheng and Liu 2005). In this
study, we measured the liver injury in mice
as the biologic end point for exploring the
mechanism of Pb-increased/LPS-induced TNF- expression.
Thus, the aims of this study were to identify
the cells in the blood responsible for TNF- release
and to reveal the role of PKC and p42/44
MAPK in the induction of TNF- during
coexposure to Pb plus LPS.
Chemicals. We obtained LPS
(derived from Escherichia coli, serotype
055:B5), TNF- inhibitor
pentoxifylline (PTX), and macrophage cytotoxic
agent GdCl3-6H2O
(gadolinium chloride hexahydrate)from Sigma
Chemical (St. Louis, MO, USA). Lead acetate
was purchased from Merck (Darmstadt, Germany).
PKC inhibitor chelerythrine chloride (C21H18NO4Cl)
was obtained from Calbiochem (Bad Soden,
Germany), and p42/44 MAPK inhibitor U0126
was purchased from Promega (Madison, WI,
USA).
Cells and animals. Peritoneal
macrophages were isolated by flushing the
peritoneal cavity of A/J mice with 10 mL
of sterile ice-cold phosphate buffer. Peritoneal
lavage fluid was centrifuged at 200g,
and pelleted cells were resuspended in Dulbecco’s
modified Eagle medium (DMEM) supplemented
with 10% fetal bovine serum (FBS), penicillin,
and streptomycin (100 U/mL). Cells were then
seeded at 2  105/well
in a 96-well plate, incubated at 37°C
for 4 hr, and washed with phosphate-buffered
saline (PBS) to remove unattached cells.
Attached cells, taken as macrophages and
confirmed with F4/80 stain, were used for
sequential experiments. RAW264.7 cells, a
mouse macrophage cell line (American Type
Culture Collection, Rockville, MD, USA),
were cultured in DMEM supplemented with 10%
FBS. A/J mice weighing 20-25 g were
obtained from and housed in the laboratory
animal center of our institution. Animals
were housed individually in a room with a
12/12-hr light/dark cycle and central air-conditioning
(25°C, 70% humidity), and were fed with
standard food ad libitum. The animal
experiment procedures were reviewed and implemented
through the Institutional Animal Care and
Use Committee process of National Cheng Kung
University. 105/well
in a 96-well plate, incubated at 37°C
for 4 hr, and washed with phosphate-buffered
saline (PBS) to remove unattached cells.
Attached cells, taken as macrophages and
confirmed with F4/80 stain, were used for
sequential experiments. RAW264.7 cells, a
mouse macrophage cell line (American Type
Culture Collection, Rockville, MD, USA),
were cultured in DMEM supplemented with 10%
FBS. A/J mice weighing 20-25 g were
obtained from and housed in the laboratory
animal center of our institution. Animals
were housed individually in a room with a
12/12-hr light/dark cycle and central air-conditioning
(25°C, 70% humidity), and were fed with
standard food ad libitum. The animal
experiment procedures were reviewed and implemented
through the Institutional Animal Care and
Use Committee process of National Cheng Kung
University.
Blood collection and biochemistry
study. The blood of mice was
collected from the inferior vena cava
under ethyl ether anesthesia, drawn using
venipuncture into serum separation tubes,
allowed to clot for 10 min at room temperature,
and then centrifuged (1,000g,
10 min, 4°C). Serum samples were
stored at 70°C. To determine the
serum concentrations of aspartate aminotransferase
(AST) and alanine aminotransferase (ALT),
serum was spotted to slides (Fuji Dri-Chem;
Fujifilm, Kanagawa, Japan) and evaluated
using a slide analyzer (Fuji Dri-Chem
3500S; Fujifilm).
Preparation of mouse whole-blood
and cytokine assays. Induction
of TNF- in
mouse whole blood was performed as described
previously (Mullarkey et al. 2003). Briefly,
Pb and LPS were added to heparinized
whole blood obtained from A/J mice (100 µL/well).
After 2 hr incubation at 37°C in
a 5% CO2 atmosphere, the blood
was centrifuged at 1,000g for
10 min at 4°C. TNF- in
conditioned medium was determined using
enzyme-linked immunosorbent assay (ELISA)
(R&D Systems, Minneapolis, MN, USA),
measuring absorbance at 450 nm and extrapolating
from a standard curve with a sensitivity
limit of 32.5 pg/mL.
Flow cytometric analysis. To
identify the TNF--secreting
cells, Pb or LPS was added to whole blood
with monensin (eBioscience, San Diego, CA,
USA). Red blood cells were lysed using hypotonic
shock, and leukocytes were subjected to surface
CD14 labeling using phycoerythrin-conjugated
CD14 antibody (eBioscience). Cells were fixed
and permeabilized using a commercial kit
(Cytofix/Cytoperm; PharMingen, San Diego,
CA, USA) and stained for intracellular TNF- using
fluorescein isothiocyanate (FITC)-conjugated
rat anti-mouse TNF- Ab
(PharMingen).To analyze the phosphorylation
status of p42/44 MAPK in peritoneal macrophages,
exudate cells were fixed using 2% formaldehyde;
they were then resuspended in methanol at
a concentration of 90%. Cells were incubated
with the primary phospho-p42/44 MAPK antibody
(New England Biolabs, Beverly, MA, USA) for
30 min at room temperature. After being washed
in PBS containing 0.5% FBS, secondary antibody
hybridization was carried out using goat
anti-rabbit IgG (Alexa Fluor 488; Molecular
Probes, Eugene, OR, USA). Macrophage-specific
marker F4/80 was first stained with biotin-conjugated
anti-F4/80 antibody and then with phycoerythrin-
conjugated streptavidin secondary antibody (eBioscience). Flow cytometric analysis
was then performed (FACSCalibur; Becton Dickinson, San Jose, CA, USA). Data
were analyzed using the CellQuest (BD Biosciences, San Jose, CA, USA)and WinMDI
2.8 software packages (University of Massachusetts, Amherst, MA, USA).
Western blot analysis. RAW264.7
cells were cultured in 0.01% FBS/DMEM for
30 min and sequentially stimulated with Pb
(10 µM), LPS (1 ng/mL), Pb (10 µM)
plus LPS (Pb+LPS) (1 ng/mL), or saline (control).
After stimulation, cells were washed with
cold PBS and then solubilized with ice-cold
buffer containing 25 mM HEPES (pH 7.5), 300
mM NaCl, 1.5 mM MgCl2, 0.2 mM
EDTA, 0.1% Triton X-100, 20 mM β-glycerophosphate,
0.1 mM sodium orthovanadate, 0.5 mM dithiothreitol,100 µg/mL
phenylmethylsulfonyl fluoride,and 2 µg/mL
leupeptin. Approximately 10-30 µg
of protein was separated using electrophoresis
in a 10% sodium dodecyl sulfate-polyacrylamide
gel. After electrophoresis, the protein was
electrotransferred onto polyvinylidene fluoride
membranes (NEN Life Science Products, Inc.,
Boston, MA, USA). Membranes were probed with
antibodies specific for phospho-p42/44 MAPK
or p42/44 MAPK (New England Biolabs). After
being probed with a horseradish peroxidase-conjugated
secondary antibody, protein signals were
visualized using enhanced chemiluminescence
reagents (Amersham, Arlington Heights, IL,
USA) combined with Kodak X-ML film exposure
(Eastman-Kodak, Rochester, NY, USA).
Histology. Liver tissue obtained
from mice was fixed in 3.7% buffered formalin
and embedded in paraffin. Sections (5 µm)
were routinely stained with hematoxylin and
eosin Y stain.
Statistical analysis. Data
are expressed as mean ± SE. We used
one-way or two-way analysis of variance and
Student’s t-test. Statistical
significance was set at p < 0.05.

Figure 1. Expression
of TNF- ( A),
AST ( B), and ALT ( C)
in A/J mice exposed to Pb, LPS,
Pb+LPS, or saline (control). ND,
not detectable. Blood was collected
1.5 hr after treatment to determine
serum TNF- ( A).
Serum AST ( B) and ALT ( C)
were evaluated 24 hr posttreatment. n =
3 per treatment.
* p < 0.05
compared with LPS.
|

Figure 2. Histologic examination
of liver damage in A/J mice challenged
with Pb (B, F, J, N, R),
LPS (C, G, K, O, S),
Pb+LPS (D, H, L, P, T),
or saline (control) (A, E, I, M, Q).
To evaluate the effects of corresponding
inhibitors, mice were pretreated with
PTX (100 mg/kg) for 60 min (E, F, G, H),
GdCl3 (40 mg/kg) for 24
hr (I, J, K, L),
or C21H18NO4Cl
(5 mg/kg) for 30 min (M, N, O, P),
or U0126 (25 µmol/kg) for 10
min (Q, R, S, T).
Mice were sacrificed after 24 hr. Arrows
in (D) indicate the necrosis
area. Tissue was stained with hematoxylin
and eosin. Bars = 0.04 mm. |

Figure 3. Effects of
TNF- inhibitor
on liver damage in A/J mice intraperitoneally
injected with PTX (100 mg/kg) or
H 2O as vehicle (control).
One hour later, mice were stimulated
with Pb, LPS, Pb+LPS, or saline
only (control). ND, not detectable.
Blood was collected either 1.5
hr after Pb, LPS, or Pb+LPS treatment
to determine serum TNF- ( A),
or 24 hr posttreatment to determine
serum AST ( B) and ALT ( C). n =
3 per treatment.
* p < 0.05
compared with Pb+LPS.
|

Figure 4. TNF- expression
in cultured whole blood of mice. Whole
blood was stimulated in vitro with
LPS (5 µg/mL) with or without
Pb acetate (1 µM) for 1.5 hr.
ND, not detectable. ( A) TNF- in
serum measured by ELISA. ( B)
Density plot for blood cells from the
Pb+LPS-treated group doubly stained
for surface CD14 and intracellular
TNF- ( x-axis,
TNF- ; y-axis,
CD14) and analyzed using flow cytometric
analysis; the percentage shown is the
ratio of TNF- + to
CD14 + cells. ( C)
Mean percentage of TNF- + cells
in CD14 + population (mean ± SE)
of groups treated with Pb, LPS, or
Pb + LPS ( n = 3–5
per treatment).
* p < 0.05
compared with LPS.
|

Figure 5. Liver damage after
inactivating monocytes/macrophages
in A/J mice intravenously injected
with GdCl 3 (40 mg/kg) or
H 2O as vehicle (control).
After 24 hr, mice were stimulated with
Pb, LPS, Pb+LPS, or saline only (control).
ND, not detectable. Blood was collected
either 1.5 hr posttreatment to determine
serum TNF- ( A)
or 24 hr posttreatment to determine
serum AST ( B) and ALT ( C). n =
3 per treatment.
* p < 0.05
compared with Pb+LPS.
|

Figure 6. P42/44 MAPK phosphorylation
in peritoneal macrophages and RAW264.7
cells. (A) Macrophages from
peritoneal exudates were verified using
F4/80 expression [x-axis, forward
scatter (FSC); y-axis, F4/80].
(B) Representative histogram
of intracellular phosphor-p42/44 MAPK
staining in F4/80high populations
showing Pb+LPS and saline. (C)
Mean percentage of phospho-p42/44+ cells
in the F4/80high population
of groups treated with Pb, LPS, or
Pb+LPS (mean ± SE, n =
3 per treatment).(D)
RAW264.7 cells stimulated with Pb (10 µM,
lane 2), LPS (1 ng/mL, lane 3), Pb
(10 µM) plus LPS (1 ng/mL, lane
4), or saline (lane 1) for 5 min; total
p42/44 MAPK and phosphorylated p42/44
MAPK were analyzed using Western blot
analysis. (E) Relative intensities
calculated by averaging three independent
experiments (± SE).
*Statistically
significant from other treatment groups
(p < 0.05).
|

Figure 7. PKC and p42/44
MAPK as inhibitors on TNF- expression
and liver damage in mice pretreated
(intraperitoneal injection) with C 21H 18NO 4Cl
(Ch; 5 mg/kg, 30 min), U0126 (25 µmol/kg,
10 min), or H 2O vehicle
(control, 30 min) and then stimulated
with Pb, LPS, Pb+LPS, or saline only
(control). ND, not detectable. Blood
was collected either 1.5 hr after treatment
to determine serum TNF- ( A),
or 24 hr posttreatment to determine
serum AST ( B) and ALT ( C). n =
3 per treatment.
* p < 0.05
Ch+Pb+LPS or U0126+Pb+LPS compared with
Pb+LPS.
|

Figure 8. PKC and p42/44
MAPK in the induction of TNF- in
peritoneal macrophages and RAW264.7
cells. ( A) TNF- measured
by ELISA in conditioned medium from
peritoneal macrophages cultured in
a 24-well plate (5 x 10 5 cells/well).
After 24 hr of attachment, cells were
stimulated with LPS (0.1 or 1 ng/mL)
combined with Pb (10 µM) for
3 hr. To evaluate the effect of kinase
inhibitors, peritoneal macrophages
were pretreated with U0126 ( B)
or C 21H 18NO 4Cl
(Ch) ( C) for 30 min and then
stimulated with 0.1 ng/mL LPS plus
10 µM Pb for 3 hr. ( D)
RAW264.7 cells were seeded in a 96-well
plate (1 x 10 4 cells/well)
and stimulated for 3 hr with Pb (0,
1, or 10 µM) combined with LPS
at various concentrations. To evaluate
the effect of kinase inhibitors, RAW264.7
cells were pretreated with U0126 ( E)
or C 21H 18NO 4Cl
( F) for 30 min and then stimulated
with 10 ng/mL LPS plus 10 µM
Pb for 3 hr. n = 3.
* p < 0.05
compared with Pb+LPS.
|
Expression of TNF- and
liver injury in mice after coexposure
to Pb plus LPS. We evaluated
the role of TNF- in
Pb-increased LPS-induced liver injury.
A/J mice were intraperitoneally given
Pb (100 µmol/kg) (Shinozuka et
al. 1996), LPS (5 mg/kg) (Ohta and Sitkovsky
2001), Pb+LPS (Pb, 100 µmol/kg;
LPS, 5 mg/kg), or saline (control). Blood
was collected 1.5 hr later to determine
serum TNF- (Figure
1A) and 24 hr later to measure the levels
of AST (Figure 1B) and ALT (Figure 1C)
that indicate liver injury. Histologic
examination of the liver was performed
24 hr posttreatment (Figure 2). Serum
TNF- was
not detected in mice that received saline
or Pb alone. Mice challenged with LPS
alone showed a small amount of serum
TNF- (< 250
pg/mL) and a slight increase in AST (300
U/L) and ALT (30 U/L). However, we observed
few histologic changes indicating hepatocellular
damage in mice that received saline (Figure
2A), Pb alone (Figure 2B), or LPS alone
(Figure 2C). Pb significantly increased
LPS-induced TNF- production
in mice treated with Pb+LPS. The induction
of serum TNF- by
LPS was drastically increased by Pb that
reached around 2,000 pg/mL in 1.5 hr.
Concurrently, the mean levels of AST
and ALT were significantly elevated in
the Pb+LPS group to 720 U/L and 600 U/L,
respectively. In addition, Pb+LPS-treated
mice showed multiple profoundly necrotic
areas in the liver (Figure 2D).
To establish a causal relationship between
TNF- and
liver injury, we used PTX, a potent inhibitor
of TNF transcription in vivo (Lechner
et al. 1993), to suppress the production
of TNF-.
Mice that had been given PTX (100 mg/kg)
1 hr before Pb+LPS treatment had lower serum
TNF- than
did those that had not been given PTX (Figure
3A). Along with a decrease in TNF- induction,
the Pb+LPS-stimulated AST (Figure 3B) and
ALT levels (Figure 3C) were also markedly
attenuated by PTX. Moreover, PTX significantly
decreased the number of necrotic hepatocellular
lesions in Pb+LPS-treated mice (Figure 2H).
TNF- producing
cells. We detected rare TNF-+ cells
in the liver using immunohistochemical
staining (data not shown). We then checked
blood cells using an in vitro whole-blood
culture model. LPS (5 µg/mL) increased
the expression of mean TNF- in
whole-blood culture to 75 pg/mL (mean
value) in 1.5 hr. Although Pb (1 µM)
itself did not induce detectable TNF-,
it significantly increased the TNF--inducing
effect of LPS, which then reached approximately
125 pg/mL (Figure 4A). Because CD14+ macrophages/monocytes
have been documented as a major source
of TNF- (Haziot
et al. 1996), we analyzed the CD14+ cells
in the whole-blood culture after Pb+LPS
coexposure. Using flow cytometric analysis,
we determined whether CD14+ cells
were the primary TNF--secreting
cells in blood. The cells were stained
with phycoerythrin-anti-CD14and
FITC-anti-TNF- after
stimulation. TNF-+/CD14+ cells
(5.9%) were separated from single negative
cells (Figure 4B, top right quadrant).
Intracellular TNF- stain
showed that < 8% of the CD14+ cells
were in untreated or Pb-treated whole-blood
culture. Approximately 12% of the CD14+ cells
in the LPS group were also TNF-+.
Coexposure to Pb+LPS increased the number
of TNF-+ cells
to about 20% of the CD14+ cells
(Figure 4C).
Macrophages/monocytes mediating Pb+LPS-induced
liver injury. To confirm whether
Pb+LPS-induced liver injury involves
macrophages/monocytes, we inactivated
macrophages/monocytes using GdCl3 (40
mg/kg) 24 hr before Pb+LPS treatment.
Mouse peritoneal macrophages that received
GdCl3 were drastically reduced,
and few cells expressed a high level
of F4/80 (F4/80high), representing
peritoneal macrophages in peritoneal
exudate (data not shown). GdCl3 decreased
the level of serum TNF- in
mice that had received Pb+LPS (Figure
5A). In parallel with a reduction in
TNF-,
GdCl3 markedly decreased serum
AST (Figure 5B) and ALT (Figure 5C) measured
24 hr after Pb+LPS treatment. GdCl3 also
reduced the number of hepatocellular
lesions in mice that received Pb+LPS,
as shown by reduced necrotic areas in
the liver (Figure 2L).
The activation of p42/44 MAPK in
peritoneal macrophages and RAW264.7 cells
treated with Pb+LPS. We examined
whether Pb acted through the common PKC
and p42/44 MAPK pathway to increase LPS-induced
TNF- expression
in peritoneal macrophages and RAW264.7
cells. The endothelial cells around large
vessels in the livers of mice treated
with Pb+LPS showed phosphorylated p42/44
MAPK, but the hepatocytes did not (data
not shown). We further characterized
the phosphorylation status of p42/44
MAPK in peritoneal macrophages, which
showed high F4/80 expression (Figure
6A). An increase in the phosphorylation
of p42/44 MAPK in peritoneal macrophages
was observed in the Pb+LPS group (Figure
6B,C). Similarly, Pb+LPS treatment induced
phosphorylation of p42/44 MAPK in RAW264.7
cells (Figure 6D,E).
Effects of PKC and p42/44 MAPK inhibitors
on TNF- and
liver injury in vivo. To
evaluate the actions of PKC and p42/44
MAPK invivo, mice were
treated with either C21H18NO4Cl
(5 mg/kg) 30 min or U0126 (25 µmol/kg)
10 min before being exposed to Pb, LPS,
or Pb+LPS. Both C21H18NO4Cl
and U0126 effectively decreased the serum
TNF- induced
by Pb+LPS (Figure 7A), as well as the
AST and ALT levels elevated by Pb+LPS
(Figure 7B,C). No obvious damage occurred
in the livers of mice that had received
C21H18NO4Cl
or U0126 alone (Figure 2M,Q). Both C21H18NO4Cl
and U0126 effectively attenuated the
necrotic lesions developed in the livers
of mice after Pb+LPS treatment (Figure
2P,T).
PKC and p42/44 MAPK in the induction
of TNF- in
peritoneal macrophages and RAW264.7 cells. Pb
(10 µM) significantly increased
the expression of TNF- induced
by low doses of LPS (0.1 and 1 ng/mL)
in mouse F4/80high peritoneal
macrophages (Figure 8A).In addition,
Pb increased the expression of TNF- in
RAW264.7 cells after treatment with 1
or 10 ng/mL LPS (Figure 8D). This Pb-induced
increase is more obvious at 10 µM
than at 1 µM.
Using MAPK and PKC inhibitors, we demonstrated
that p42/44 MAPK and PKC were involved in
the Pb+LPS-induced TNF- expression
of peritoneal macrophages and RAW264.7 cells.
At doses between 7.5 and 30 µM, U0126,
a p42/44 MAPK inhibitor, significantly suppressed
the Pb+LPS-induced expression of TNF-.
(Figure 8B,E). At a dose of 5 µg/mL,
C21H18NO4Cl,
a PKC inhibitor, also reduced Pb+LPS-associated
TNF- expression
(Figure 8C,F).
Monocytes/macrophages are the primary secretors
of TNF- during
inflammation and infection (Beutler and Cerami
1989). We demonstrated that the harmful effects
of these cells triggered by LPS may become
worse for the host in the presence of Pb,
as reflected by severe liver injury. Inactivating
the function of these phagocytic cells and
blocking the signal pathways for TNF- production
effectively relieve the damage caused by
environmental insults such as LPS and Pb.
Pb and low doses of LPS neither directly
stimulated TNF-+ production
nor activated phosphorylation of MAPK in
hepatocytes other than endothelial cells.
Previous studies on the cellular source of
TNF- in
animal exposed to Pb or LPS were not conclusive.
Pb might increase the transcription of TNF- mRNA
in hepatocytes (Kubo et al. 1996), and LPS
might stimulate liver Kupffer cells to release
TNF- (Suzuki
et al. 1996; Tsukada et al. 2003). However,
in a mixed culture of hepatocytes and Kupffer
cells, Pb and LPS stimulated only a small
increase in the production of TNF- that
did not cause obvious cell death in the cultured
hepatocytes (Milosevic and Maier 2000; Zhang
et al. 1996). In our animal model, serum
TNF- rapidly
(within 1.5 hr) reached maximal levels after
the Pb+LPS challenge. Moreover, cells releasing
TNF- after
LPS stimulation in vivo have been
identified as CD14+ (Haziot et
al. 1996; Perera et al. 1997), but Kupffer
cells express little CD14 (Lichtman et al.
1998). We also found that the TNF- induced
by Pb+LPS was expressed primarily by CD14+ cells
in the whole-blood culture and in mouse F4/80+ peritoneal
macrophages, which indicates that the TNF- was
produced by cells outside the liver. The
increase in TNF- caused
by Pb in the serum of LPS-treated mice was
approximately 10-fold. However, the increase
of TNF- was
only 2-fold in peritoneal macrophages. This
indicates that other cells and mechanisms
might contribute to the Pb-increased LPS-induced
TNF- production in
vivo and need further investigation.
A second line of evidence excluding hepatocytes
as the source of TNF- comes
from studies of cell signaling. PKC and p42/44
MAPK are downstream signals of Pb stimulation
in neurons (Olivi et al. 2003), bone-marrow-derived
macrophages (Flohe et al. 2002), and glioma
cells (Cheng et al. 2004), and they may regulate
genes responding to Pb poisoning. Long-term
exposure to Pb leads to PKC activation in
rat livers; however, Pb also inhibited the
activity of PKC in
a human hepatoma cell line (Liu et al. 1997;
Tonner and Heiman 1997), and the inhibition
was much more pronounced when Pb levels were
high (Sun et al. 1999). Because circulating
Pb will quickly deposit in the liver (Bornemann
and Colburn 1985), we speculated (Cheng and
Liu 2005) that a transient accumulation of
Pb in the liver will suppress the activation
of PKC and p42/44 MAPK in hepatocytes. Pb
or LPS alone at the doses we used did not
alter the phosphorylation of p42/44 MAPK
in HepG2 cells (data not shown). In addition,
in our animal model, we did not find phosphorylation
of p42/44 MAPK in hepatocytes, further indicating
that they were not induced to produce TNF- in
response to Pb exposure. In contrast to liver
cells, coexposure to Pb+LPS significantly
stimulated p42/44 MAPK phosphorylation in
peritoneal macrophages; suppressing p42/44
MAPK phosphorylation or inhibiting PKC activity
resulted in reduced TNF- expression.
As mentioned above, both Pb and a high
dose of LPS activated PKC and MAPK, which
stimulated TNF- expression
(Cheng et al. 2004). In our study model,
the synergistic effect of Pb+LPS was demonstrable:
coexposing mice to Pb+LPS strongly induced
TNF- expression
in peritoneal macrophages and in whole blood.
In addition, significant interaction between
the LPS-treatment factor and Pb-dose factor
was indicated by dose-dependent TNF- expression
in different groups of RAW264.7 cells. Although
Pb alone or LPS alone slightly increased
the phosphorylation of MAPK in peritoneal
macrophages, only coexposing them to Pb+LPS
induced obvious p42/44 MAPK phosphorylation
and TNF- expression.
Apparently, the minimal level of MAPK activation
required to induce a large amount of TNF- was
higher than the levels of MAPK in cells after
exposure to a single dose of Pb alone or
LPS alone. After the signals initiated by
Pb+LPS merged in the PKC and MAPK pathway,
MAPK activation was sufficient for TNF- production.
That an otherwise insignificant effect of
LPS in mice became vital after the mice had
been coexposed to Pb+LPS has implications
for setting the maximal tolerable concentration
for a particular pollutant. It seems that
risk assessment based on a single-exposure
experimental design does not truly reflect
the hazard of an environmental pollutant
to humans.
In conclusion, the PKC/MAPK pathway leading
to TNF- expression
played a key role in Pb+LPS-induced liver
injury in mice. Our results also indicate
that immune cells are very sensitive to environmental
pollution and emphasize the synergistic effect
of multiple pollutants in disease progression.
Specifically, monocytes/macrophages may serve
as watchful janitors in response to Pb+LPS,
even when it injures their host. |
|
|
| [References Listed in PubMed] References
Aggarwal BB. 2003. Signalling pathways of the TNF superfamily: a double-edged sword. Nat Rev Immunol 3(9):745-756. [CrossRef].
Beutler B, Cerami A. 1989. The biology of cachectin/TNF--a primary mediator of the host response. Annu Rev Immunol 7:625-655.
Bornemann LD, Colburn WA. 1985. Pharmacokinetic model to describe the disposition of lead in the rat. J Toxicol Environ Health 16(3-4):631-639.
Cheng YJ, Liu MY. 2005. Modulation of tumor necrosis factor-alpha and oxidative stress through protein kinase C and P42/44 mitogen-activated protein kinase in lead increases lipopolysaccharide-induced liver damage in rats. Shock 24(2):188-193.
Cheng YJ, Liu MY, Wu TP, Yang BC. 2004. Regulation of tumor necrosis factor-alpha in glioma cells by lead and lipopoly-saccharide: involvement of common signaling pathway. Toxicol Lett 152(2):127-137.
Comalada M, Xaus J, Valledor AF, Lopez-Lopez C, Pennington DJ, Celada A. 2003. PKC epsilon is involved in JNK activation that mediates LPS-induced TNF-alpha, which induces apoptosis in macrophages. Am J Physiol Cell Physiol 285(5):C1235-C1245.
Costa LG. 1998. Signal transduction in environmental neurotoxicity. Annu Rev Pharmacol Toxicol 38:21-43. [CrossRef].
Dentener MA, Greve JW, Maessen JG, Buurman WA. 1989. Role of tumour necrosis factor in the enhanced sensitivity of mice to endotoxin after exposure to lead. Immunopharmacol Immunotoxicol 11(2-3):321-334.
Flohe SB, Bruggemann J, Herder C, Goebel C, Kolb H. 2002. Enhanced proinflammatory response to endotoxin after priming of macrophages with lead ions. J Leukoc Biol 71(3):417-424.
Ganey PE, Roth RA. 2001. Concurrent inflammation as a determinant of susceptibility to toxicity from xenobiotic agents. Toxicology 169(3):195-208.
Gao HM, Jiang J, Wilson B, Zhang W, Hong JS, Liu B. 2002. Microglial activation-mediated delayed and progressive degeneration of rat nigral dopaminergic neurons: relevance to Parkinson’s disease. J Neurochem 81(6):1285-1297.
Goldfeld AE, Doyle C, Maniatis T. 1990. Human tumor necrosis factor alpha gene regulation by virus and lipopolysaccharide. Proc Natl Acad Sci USA 87(24):9769-9773.
Goldstein GW. 1993. Evidence that lead acts as a calcium substitute in second messenger metabolism. Neurotoxicology 14(2-3):97-101.
Haziot A, Ferrero E, Kontgen F, Hijiya N, Yamamoto S, Silver J, et al. 1996. Resistance to endotoxin shock and reduced dissemination of gram-negative bacteria in CD14-deficient mice. Immunity 4(4):407-414.
Hewett JA, Jean PA, Kunkel SL, Roth RA. 1993. Relationship between tumor necrosis factor-alpha and neutrophils in endotoxin-induced liver injury. Am J Physiol 265(6 Pt 1):G1011-G1015.
Kosai K, Matsumoto K, Funakoshi H, Nakamura T. 1999. Hepatocyte growth factor prevents endotoxin-induced lethal hepatic failure in mice. Hepatology 30(1):151-159.
Kubo Y, Yasunaga M, Masuhara M, Terai S, Nakamura T, Okita K. 1996. Hepatocyte proliferation induced in rats by lead nitrate is suppressed by several tumor necrosis factor alpha inhibitors. Hepatology 23(1):104-114.
Kunstle G, Leist M, Uhlig S, Revesz L, Feifel R, MacKenzie A, et al. 1997. ICE-protease inhibitors block murine liver injury and apoptosis caused by CD95 or by TNF-alpha. Immunol Lett 55(1):5-10.
Lechner AJ, Rouben LR, Potthoff LH, Tredway TL, Matuschak GM. 1993. Effects of pentoxifylline on tumor necrosis factor production and survival during lethal E. coli sepsis vs. disseminated candidiasis with fungal septic shock. Circ Shock 39(4):306-315.
Leist M, Gantner F, Kunstle G, Bohlinger I, Tiegs G, Bluethmann H, et al. 1996. The 55-kD tumor necrosis factor receptor and CD95 independently signal murine hepatocyte apoptosis and subsequent liver failure. Mol Med 2(1):109-124.
Lichtman SN, Wang J, Lemasters JJ. 1998. LPS receptor CD14 participates in release of TNF-alpha in RAW264.7 and peritoneal cells but not in Kupffer cells. Am J Physiol 275(1 Pt 1):G39-G46.
Liu JY, Lin JK, Liu CC, Chen WK, Liu CP, Wang CJ, et al. 1997. Augmentation of protein kinase C activity and liver cell proliferation in lead nitrate-treated rats. Biochem Mol Biol Int 43(2):355-364.
Liu MY, Cheng YJ, Chen CK, Yang BC. 2005. Coexposure of lead- and lipopolysaccharide-induced liver injury in rats: involvement of nitric oxide-initiated oxidative stress and TNF-alpha. Shock 23(4):360-364.
Liu MY, Lai HY, Yang BC, Tsai ML, Yang HY, Huang BM. 2001. The inhibitory effects of lead on steroidogenesis in MA-10 mouse Leydig tumor cells. Life Sci 68(8):849-859.
Luster MI, Faith RE, Kimmel CA. 1978. Depression of humoral immunity in rats following chronic developmental lead exposure. J Environ Pathol Toxicol 1(4):397-402.
Markovac J, Goldstein GW. 1988. Lead activates protein kinase C in immature rat brain microvessels. Toxicol Appl Pharmacol 96(1):14-23.
Milosevic N, Maier P. 2000. Lead stimulates intercellular signalling between hepatocytes and Kupffer cells. Eur J Pharmacol 401(3):317-328.
Mullarkey M, Rose JR, Bristol J, Kawata T, Kimura A, Kobayashi S, et al. 2003. Inhibition of endotoxin response by e5564, a novel Toll-like receptor 4-directed endotoxin antagonist. J Pharmacol Exp Ther 304(3):1093-1102.
Ohta A, Sitkovsky M. 2001. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature 414(6866):916-920.
Olivi L, Sisk J, Bressler J. 2003. The involvement of lipid activators of protein kinase C in the induction of ZIF268 in PC12 cells exposed to lead. Neurochem Res 28(1):65-71.
Perera PY, Vogel SN, Detore GR, Haziot A, Goyert SM. 1997. CD14-dependent and CD14-independent signaling pathways in murine macrophages from normal and CD14 knockout mice stimulated with lipopolysaccharide or taxol. J Immunol 158(9):4422-4429.
Pfeffer K. 2003. Biological functions of tumor necrosis factor cytokines and their receptors. Cytokine Growth Factor Rev 14(3-4):185-191. [CrossRef].
Pounds JG. 1984. Effect of lead intoxication on calcium homeostasis and calcium-mediated cell function: a review. Neurotoxicology 5(3):295-331.
Rosen JF, Pounds JG. 1989. Quantitative interactions between Pb2+ and Ca2+ homeostasis in cultured osteoclastic bone cells. Toxicol Appl Pharmacol 98(3):530-543.
Schanne FA, Dowd TL, Gupta RK, Rosen JF. 1989. Lead increases free Ca2+ concentration in cultured osteoblastic bone cells: simultaneous detection of intracellular free Pb2+ by 19F NMR. Proc Natl Acad Sci USA 86(13):5133-5135.
Schuchmann M, Varfolomeev EE, Hermann F, Rueckert F, Strand D, Koehler H, et al. 2003. Dominant negative MORT1/FADD rescues mice from CD95 and TNF-induced liver failure. Hepatology 37(1):129-135.
Selye H, Tuchweber B, Bertok L. 1966. Effect of lead acetate on the susceptibility of rats to bacterial endotoxins. J Bacteriol 91(2):884-890.
Shinozuka H, Ohmura T, Katyal SL, Zedda AI, Ledda-Columbano GM, Columbano A. 1996. Possible roles of nonparenchymal cells in hepatocyte proliferation induced by lead nitrate and by tumor necrosis factor alpha. Hepatology 23(6):1572-1577.
Spooner CE, Markowitz NP, Saravolatz LD. 1992. The role of tumor necrosis factor in sepsis. Clin Immunol Immunopathol 62(1 Pt 2):S11-S17.
Sun X, Tian X, Tomsig JL, Suszkiw JB. 1999. Analysis of differential effects of Pb2+ on protein kinase C isozymes. Toxicol Appl Pharmacol 156(1):40-45.
Suzuki S, Nakamura S, Serizawa A, Sakaguchi T, Konno H, Muro H, et al. 1996. Role of Kupffer cells and the spleen in modulation of endotoxin-induced liver injury after partial hepatectomy. Hepatology 24(1):219-225.
Tonner LE, Heiman AS. 1997. Lead may affect glucocorticoid signal transduction in cultured hepatoma cells through inhibition of protein kinase C. Toxicology 119(2):155-166.
Triantafilou M, Triantafilou K. 2002. Lipopolysaccharide recognition: CD14, TLRs and the LPS-activation cluster. Trends Immunol 23(6):301-304. [CrossRef].
Tsukada S, Enomoto N, Takei Y, Hirose M, Ikejima K, Kitamura T, et al. 2003. Dalteparin sodium prevents liver injury due to lipopolysaccharide in rat through suppression of tumor necrosis factor-alpha production by Kupffer cells. Alcohol Clin Exp Res 27(8 suppl):7S-11S.
Ulich TR, Watson LR, Yin SM, Guo KZ, Wang P, Thang H, et al. 1991. The intratracheal administration of endotoxin and cytokines. I. Characterization of LPS-induced IL-1 and TNF mRNA expression and the LPS-, IL-1-, and TNF-induced inflammatory infiltrate. Am J Pathol 138(6):1485-1496.
Zhang F, Warskulat U, Haussinger D. 1996. Modulation of tumor necrosis factor-alpha release by anisoosmolarity and betaine in rat liver macrophages (Kupffer cells). FEBS Lett 391(3):293-296.
Last Updated: April 20, 2006
|
|
|
|
| |










