Free-radical reactions dominate the combustion process of organic materials. Primary combustion radicals are reactive species and most have lifetimes in air of less than a microsecond. Despite their reactivity, free radicals have been detected in cigarette smoke as long as 5 min after combustion (
1-3). Studies have shown that the radicals detected in aged cigarette smoke and other postcombustion gases are not remnants of the actual combustion process, but rather are formed within smoke plumes via dynamic processes involving reactions of species such as NO, NO
2, O
2, and incompletely oxidized organic compounds (
2-5). Because radicals are formed within the aging smoke, they can be biologically important when smoke is inhaled, and can play an important role in destroying and forming other organic compounds in the smoke.
In this work we demonstrate a method to determine concentrations of each of a suite of carbon-centered radicals trapped from smoke samples--effectively a radical "fingerprint" of the sample. We then apply the method to compare radical populations in several tobacco smoke samples to those trapped from model systems comprised of isoprene and nitric oxide. We also show that actual measured radical concentrations in smoke represent a substantial underestimate (~ 100-fold); these are likely higher than estimated in previous work because of competitive scavenging of these reactive species by oxygen.
Inhaled radicals produce adducts with DNA that lead to mutations and/or cancer (6) and are likely contributors to many of the negative health effects of tobacco smoke in lungs. Radicals are also suspected agents in the sometimes mysterious smoke inhalation deaths that do not appear to result from major smoke constituents such as carbon monoxide (7-9). The most damaging radical reactions are believed to involve the alveolar lining of the lung, where reactions with lipids and proteins cause immediate alterations to the lung's ability to take up oxygen (9). The damaging radicals may include inorganic (NO2,  OH, OOH), carbon-centered organic (R), or oxygen-centered organic (OR, OOR) species. Presumably, the initial organic radicals formed are carbon-centered, and they react with O2 to generate a myriad of oxygen-centered radical compounds. Thus, detection of carbon radicals present in smoke is a critical step toward understanding the overall radical chemistry occurring in the system.
OH, OOH), carbon-centered organic (R), or oxygen-centered organic (OR, OOR) species. Presumably, the initial organic radicals formed are carbon-centered, and they react with O2 to generate a myriad of oxygen-centered radical compounds. Thus, detection of carbon radicals present in smoke is a critical step toward understanding the overall radical chemistry occurring in the system.
Development of a working model for radical formation, based on major smoke constituents, is essential for understanding the system better. Several different reaction mechanisms can explain the production of carbon-centered radicals in postcombustion gases. Continuous reaction of hydrocarbons, especially alkenes, with NO2 and OH--both of which are produced through reactions of nitric oxide within aging smoke--forms carbon-centered radicals. Additionally, peroxy radicals (OOR), which are also produced as the smoke ages, react through a series of steps to produce more radicals, including NO2 and OH. Thus, a basic model system should include NO and O2 (which react to produce NO2) and an alkene found in tobacco smoke. Over 4,000 chemical species have been identified in gas-phase cigarette smoke and many are possible participants in radical-generating reactions; several of the most abundant compounds are listed in Table 1.
Table 1. Abundant chemical species found in gas-phase cigarette smoke (5).
|
|
Compound
|
ppm
|
|
|
Nitric oxide (NO)
|
700
|
|
Isoprene
|
410
|
|
Methanol
|
390
|
|
Acetaldehyde
|
1,430
|
|
Water
|
15,500
|
|
Several researchers, using spin trapping methods and/or infrared (IR) spectroscopy, have attempted to use simple gas-phase mixtures of the most prevalent smoke compounds to model free-radical formation in cigarette smoke (2-5,10). The most successful smoke modeling to date was reported by Cueto and Pryor (5), who used IR to monitor the progression of NO and NO2 in the presence of NO, air, isoprene, and methanol. Using these same model compounds, Pryor et al. (2) compared electron paramagnetic resonance (EPR) spectra collected from spin trapping of radicals in cigarette and model systems. Although these studies have all elucidated pieces of the radical production puzzle, none have directly compared trapped radical suites from cigarette smoke and model systems. The radical detection method described in this paper is specific for carbon-centered radicals, and thus focuses on an important subset of the total radical population. The objectives of this work are to compare the suites of carbon-centered radicals trapped from different types of tobacco smoke and from previously developed model systems.
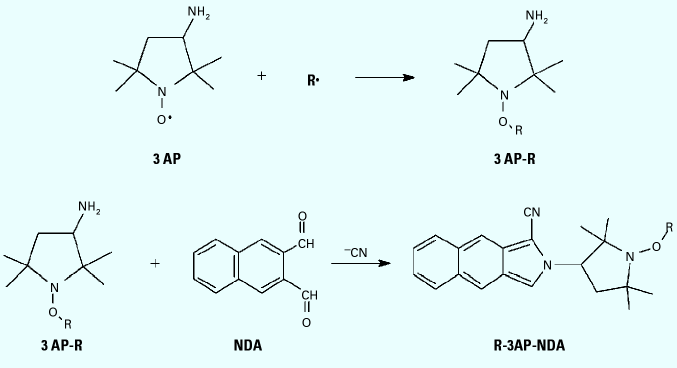
We recently reported on a novel method to trap and separate carbon-centered radicals from gas-phase mixtures such as cigarette smoke and diesel exhaust (11). The method employs a nitroxide probe [3-amino-2,2,5,5-tetramethyl-1-pyrrolidinyloxy (3AP)] on a solid-phase support to trap radicals directly from the gas phase. This technique avoids complications associated with the previously employed spin trap methods which required the sample gas to be bubbled through a solution. 3AP molecules bond covalently with carbon-centered radicals (R) to form an oxygen-carbon bond and produce the stable adducts radical-3AP adduct (R-3AP) (Figure 1). The set of adducts is then derivatized in solution with naphthalenedicarboxaldehyde (NDA), through the amino group on 3AP, to give a fluorescent product, radical-3AP adduct, NDA derivative (R-3AP-NDA). Fluorescence intensity of the resulting solution is correlated with the number of radicals trapped. The full benefits of the method are realized with mixtures of radicals because the resulting fluorescent products can be separated by HPLC to give concentrations of individual species (12,13), a procedure not possible with standard spin traps. By applying this method to the detection of radical suites from tobacco smoke and model systems, we can evaluate the models further and draw conclusions on their relevance.
Figure 1. Trapping reaction of nitroxide (3AP) with carbon-centered radical (R), followed by solution-phase derivatization with NDA to produce the fluorescent species R-3AP-NDA.
Chemicals. We purchased sodium cyanide, 3AP, NDA, cyclopentylamine (CPA), isoprene, and HPLC grade methanol from Acros (Pittsburgh, PA); sodium borate from Fisher Scientific (Pittsburgh, PA); 1,000 parts per million (ppm) NO as a reference standard from Praxair (Bethlehem, PA); and hydrocarbon-free compressed air from Praxair (Chicago, IL). All chemicals were of the highest purity available and were used without further purification. We purchased Marlboro 100 cigarettes, Djarum Special clove cigarettes, and Swisher Sweets Slim cigars from a local vendor and used them without further preparation; tobacco products were stored in the freezer and used within one month. All water used for experiments was from a Millipore Milli-Q system (Bedford, MA). Five mM NDA in methanol was prepared weekly and stored in the freezer at -5°C. Aqueous 10 mM sodium cyanide solutions were prepared monthly and stored at room temperature. We made 25 mM sodium borate buffer (pH 9.1) from Na2B4O7·10H2O and adjusted the pH with 1 M NaOH.
Procedure for trap loading. The method for coating 3AP onto glass beads was described in a previous publication (11). The beads used for these experiments were 3 mm diameter solid glass beads (Fisher Scientific) instead of the 6 mm beads used in the earlier report. We loaded the trapping compound (3AP) onto the beads by dissolving 3AP into a small volume of acetone, adding the solution to the beads in a round-bottom flask, and drying (via slow rotary evaporation) at 23 ± 3°C. For gas-phase sampling, we loaded the 3AP-coated glass beads into a standard 15 cm distillation column with a small ball of wire as a plug to hold the beads in place (Figure 2), and passed the gas to be sampled through the column.
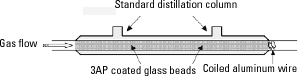
Figure 2. Apparatus used to trap carbon-centered radicals from the gas phase.
Procedure for surface area comparisons. To test for the effects of bead surface area, we used a combination of two bead sizes in the sampling columns: a) 6 mm diameter beads gave a total trapping surface area of 79 cm2; b) 3 mm diameter beads gave an area of 149 cm2; and c) a mixture of the two sizes gave an area of 112 cm2. In this experiment the standard cigarette sampling method was used with Marlboro cigarettes.
Procedure for tobacco smoke sampling. We used the standard puff protocol, described elsewhere (1), to sample tobacco smoke from Marlboro 100s and Djarum cigarettes and Swisher Sweets cigars. We took samples with a 4-way flow splitter to allow three samples and a blank (no 3AP) to be collected simultaneously on four individual columns. This improved sampling method required the use of 45 mL puffs to completely fill all four sampling columns (air volume 10 mL each) with each puff. Marlboro 100s and Djarum cigarette smoke samples consisted, respectively, of seven and four filtered cigarettes, smoked sequentially. Because of the size and nature of cigars, these samples consisted of only three cigars. Smoke was not filtered by external means, only by the manufacturer's filter, if present. Measurements made with a Digi-Sense Thermometer (Cole Parmer, Vernon Hills, IL) showed that the smoke attained room temperature within seconds during the puff sampling, so no cooling of the columns was required. The following volumes of smoke were sampled per column: 1,300 mL Marlboro cigarette smoke, 1,300 mL Djarum cigarette smoke, and 2,500 mL Swisher Sweet cigar smoke.
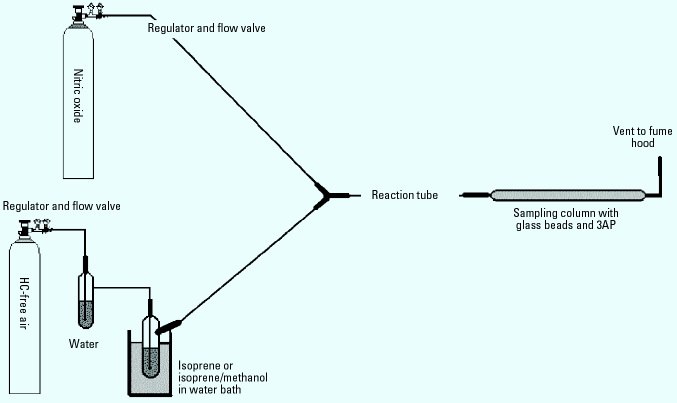
Procedure for model system studies. The model systems for cigarette smoke employed an apparatus and procedure modified from literature reports (14). Figure 3 shows a schematic of the system. The 0.25-inch ID tubing and fittings used in the entire system were made of Kynar from Cole-Parmer Company (Vernon Hills, IL). We chose Kynar for its excellent chemical resistance, low gas-permeability, and low cost. Both the lengths of the reaction coil and the gas flow rates through the system could be changed to allow for different reaction times. We used 1 m, 4 m, and 5 m tubes to determine the optimal reaction time in the model systems. Lower flow rates of the gases through the system produced longer residence/reaction times.
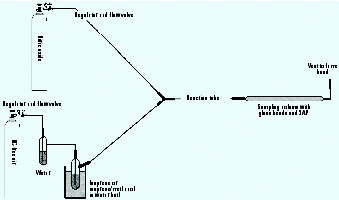
Figure 3. Diagram of model system apparatus. Compressed hydrocarbon-free air and NO in N2 are used as carrier gases. Air is first bubbled through water and then over liquid isoprene or isoprene/methanol in a water bath. The two gas streams are mixed and run through the reaction tube (1, 4, or 5 m) before sampling for radicals.
We purchased nitric oxide at a fixed concentration of 1,000 ppm in N2, and added it to the model system through a metering valve. Hydrocarbon-free air was bubbled through water to saturate the air stream and mimic the high humidity of tobacco smoke, and then blown across the surface of methanol and/or isoprene before being mixed with the NO stream. Isoprene concentrations within the reaction tube were 590 ppm (± 50); methanol concentrations were 85 ppm (± 3) as determined by gas chromatography (GC). NO concentrations were approximately 500 ppm in the reaction tube. Unless otherwise noted, we set flow rates of each of the two gases at 100 mL/min (± 10 mL/min) using a bubble flowmeter and the regulator valves. We collected samples using the four-column sampler for 20 min; thus approximately 1,000 mL of gas was sampled per column.
We observed a light yellow film inside the tubing and on the sample beads after sampling the model systems. Cueto and Pryor (5) recorded a similar phenomenon and reported that the yellow compound had Fourier-transform infrared spectroscopy (FT-IR)-based assignments from alkyl nitrates, nitroalkanes, and primary nitro- compounds. After sampling, the reaction tube was rinsed sequentially with acetone, acetonitrile, and water to remove the yellow residue. The tubes were then flushed with a high flow rate of air until dry.
Procedure for isoprene and methanol calibration. Concentrations of gas-phase isoprene and methanol in the model systems were determined by gas chromatography (GC). Isoprene and methanol standards were made by adding known small volumes (1-50 µL) of the pure liquids to 40 mL I-Chem Certified 200 Series (Fisher Scientific) amber glass vials. The compounds volatilized completely in the vials, and we generated linear calibration curves using 10 mL gas injections of these standards run on a Hewlett-Packard 6890 GC-FID (Hewlett-Packard, Wilmington, DE). We used triplicate injections of each of the 4 standards, with concentrations from 170-850 ppm isoprene and 20-400 ppm methanol, for the calibration curves (r2 = 0.99). Outflow from the model systems (with no NO present) was flushed through the I-Chem vials for 1 min, the vials were capped, and the vial headspace was sampled by the same GC method described above.
Blanks and controls. We generated two types of blanks for each system. The first type was identical to the real samples except no 3AP was placed on the beads; this blank was collected in parallel to real samples in a column with uncoated glass beads (no 3AP). This procedure would show fluorescent HPLC peaks for any primary amines that were retained on the beads and subsequently derivatized by NDA. The second type of blank was identical to the standard sampling method, except there was no NDA derivatization step. We collected this sample from the wash of any of the sampling columns. This second blank would show any interfering fluorescent species from the smoke that partitioned to the 3AP-coated glass beads.
NDA derivatization procedure. The procedure for derivatization of 3AP is described extensively in our earlier report (11) on this work and has been modified only slightly. After collecting samples, we washed the glass beads with borate buffer solution (5 mL), and derivatized an aliquot (500 µL) of this solution with a 1.5 M excess of NDA and cyanide. After 1 hr we filtered the derivatized sample with a 0.2 µm syringe filter concentrating the product as a solid on the filter. We then washed it from the filter with 20% water/80% methanol (1 mL). The procedure was improved for this study by the addition of a second precipitation step: We added 1 mL of buffer to the first filtrate, which was then re-filtered and again rinsed with the water/methanol solution (1 mL); the two filter rinse solutions were combined. The overall procedure effectively concentrated the sample and was 99% efficient in recovering 3AP-NDA species (based on absorbance).
Fluorescence calibration standards. We used cyclopentylamine-NDA (CPA-NDA) for calibration because it was easy to make and has a similar structure to that of 3AP-NDA. We derivatized CPA using the same procedure described above for 3AP. The CPA-NDA reaction is very rapid and was complete in less than 15 min. For the HPLC calibrations we assumed that R-3AP-NDA had a similar fluorescence quantum yield to that of CPA-NDA; errors associated with this assumption are likely estimated to be less than 10%.
Instrumentation
HPLC. The HPLC system was a Beckman System Gold (Fullerton, CA) with Model 126 solvent module (quaternary pump), Model 508 autosampler, and Model 168 diode-array absorbance detector. A Phenomenex ThermaSphere Model TS-430 column chiller/heater (Phenomenex, Torrance, CA) was used to maintain constant temperature. The fluorescence detector was a Jasco (Tokyo, Japan) Model FP-920. We used Beckman System Gold for Windows software for data acquisition and processing. We used a reverse-phase alkyl-amide column [250  2.1 mm, 5 µm ABZ+Plus (Supelco, Bellefonte, PA)] for all separations. The fluorescence detector was set to 420 nm excitation and 480 nm emission, corresponding to the NDA fluorophore. We used gain settings of 10 or 100 on the fluorescence detector. We performed separations at 25°C, with a flow rate of 0.3 mL/min. The mobile phase consisted of a gradient from 30% water/70% methanol to 20% water/80% methanol over 20 min. We ran CPA-NDA calibration standards with an isocratic 20% water/80% methanol mobile phase, which eluted the compound in less than 10 min. We used nine CPA-NDA standards to generate two calibration curves (one for each detector setting, gain = 10 and 100) as described above. Injections ranged from 5 to 250 pmol CPA-NDA (with injection volumes of 5 to 50 µL) and both plots were linear (r = 0.999) over the appropriate range. Peaks for each chromatogram were integrated using the standard Beckman System Gold integration software. We used the sum of the integrated peak areas from each sample, minus any background from the blanks, to calculate the number of radicals trapped.
2.1 mm, 5 µm ABZ+Plus (Supelco, Bellefonte, PA)] for all separations. The fluorescence detector was set to 420 nm excitation and 480 nm emission, corresponding to the NDA fluorophore. We used gain settings of 10 or 100 on the fluorescence detector. We performed separations at 25°C, with a flow rate of 0.3 mL/min. The mobile phase consisted of a gradient from 30% water/70% methanol to 20% water/80% methanol over 20 min. We ran CPA-NDA calibration standards with an isocratic 20% water/80% methanol mobile phase, which eluted the compound in less than 10 min. We used nine CPA-NDA standards to generate two calibration curves (one for each detector setting, gain = 10 and 100) as described above. Injections ranged from 5 to 250 pmol CPA-NDA (with injection volumes of 5 to 50 µL) and both plots were linear (r = 0.999) over the appropriate range. Peaks for each chromatogram were integrated using the standard Beckman System Gold integration software. We used the sum of the integrated peak areas from each sample, minus any background from the blanks, to calculate the number of radicals trapped.
Gas chromatography. The gas chromatograph used for isoprene and methanol calibrations was a Hewlett-Packard 6890 GC-FID. We used a diphenyl-dimethyl polysiloxane phase capillary column [DB5-MS 30 m, 0.25 mm I.D., 0.5 µm film (J&W Scientific, Folsom, CA)] for all analysis. We performed 10 µL gas injections with a 10 µL gas-tight glass syringe. Operating conditions for the GC (oven temperature 40°C; injector, 100°C; detector, 300°C) were selected for optimal chromatography and minimal elution times.
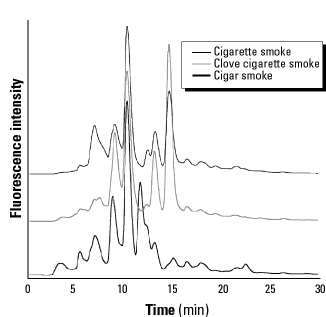
Tobacco smoke. Representative chromatograms resulting from puff-sampling smoke from Marlboro 100s cigarettes, Djurum Special clove cigarettes, and Swisher Sweet cigars are shown in Figure 4. Each fluorescent peak in the chromatogram corresponds to a different trapped carbon-centered radical (12,13). The cigarette chromatogram is very similar to that which we published previously (11), and results were reproducible from day to day. Appropriate blanks, both with and without 3AP, produced only minor background fluorescent peaks.
|
Figure 4. Representative fluorescence chromatograms of the carbon-centered radical suite collected from varieties of smoke with the standard puff method of sampling. Each peak represents a different trapped radical.
|
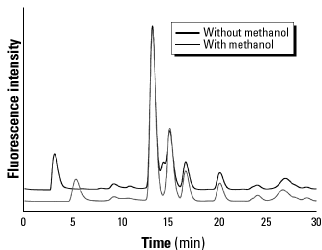
Model system. Representative chromatograms from sampling 20 min of the isoprene-air-NO model system (after ~10 sec reaction time) both with and without methanol are shown in Figure 5. We placed the sampling columns directly at the output of the 1 m reaction tube and passed the reactive gas mixture through the columns. These chromatograms were fairly reproducible from day to day; but compared with the tobacco samples, they seemed much more sensitive to factors such as the age or cleanliness of the reaction tubes, humidity, concentrations, or unknown parameters. Again, each fluorescent peak in the chromatogram corresponds to a different trapped carbon-centered radical; appropriate blanks produced only minor background fluorescence.
|
Figure 5. Representative fluorescence chromatograms of the radical suite collected from a 20-min sampling of the air, isoprene (590 ppm), NO (500 ppm) model system with 1-m reaction tube.
|
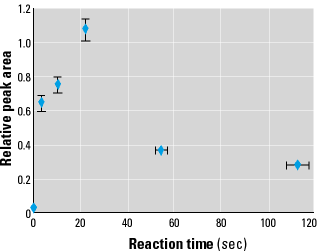
Reaction times. Figure 6 illustrates a plot of total integrated fluorescence intensity (directly related to number of radicals trapped) versus reaction time for the model system. We varied the reaction time by changing the reaction coil length and the gas flow rate, with reaction time error values estimated at ± 5%. Fluorescence peak areas for each reaction time were integrated over the entire chromatogram and normalized to the sample with the largest area (22 sec sample). Each point represents triplicate samples.
|
Figure 6. Plot of integrated fluorescence intensity (relative peak area) versus reaction time for the isoprene/air/NO model system. Reaction time error bars represent the estimated 10% uncertainty. Peak area error bars represent one standard deviation.
|
Radical quantities. Table 2 lists the numbers of radicals trapped from each of the different systems. Concentrations are listed both as nanomoles per tobacco unit (cigarette or cigar) and as picomoles per milliliter of gas sampled (at standard temperature and pressure).
Table 2. Numbers of carbon-centered radicals (R) trapped from each system.
|
|
System
|
Nanomole R trapped/cig
|
Picomole R/mL gas
|
|
|
Cigarette
|
54 ± 2
|
40 ± 2
|
|
Clove cigarette
|
66 ± 9
|
50 ± 7
|
|
Cigar
|
185 ± 9
|
73 ± 4
|
|
Isoprene + air + NO model
|
-
|
18 ± 3
|
|
Isoprene + air + methanol + NO model
|
-
|
15 ± 2
|
Cig, cigarettes and cigars.
|
Comparison of the radical suites collected from cigarettes, clove cigarettes, and cigars shows similarities and differences among radical populations in the three tobacco samples (Figure 4). In the chromatograms, each peak corresponds to an individual trapped radical, and the peak heights indicate relative concentrations. Each tobacco type produced a unique radical suite, which can be considered a fingerprint of the smoke under the given combustion conditions. At least 15 different peaks are apparent, with varying intensities for the three samples. The most dominant peaks eluted at 10 and 15 min; these peaks can reasonably be assumed to correspond to the same pair of radicals in each sample. The ratios between these two peak heights (10-min peak/15-min peak) were dramatically different for the three tobaccos: 1.8 for cigarettes, 0.8 for clove cigarettes, and about 20 for cigars. Although cigar smoke had only a small shoulder peak at 15 min, it had a large peak at 12 min which was absent in the other samples. All samples showed substantial peaks at 9 min and others of varying intensity at 5, 7, and 13 min, as well as a trail of smaller peaks between 15 and 25 min. Most of the fluorescent peaks eluted between 5 and 15 min for all three tobacco types; by comparison, C(O)CH3 adducts elute at approximately 15 min, and CH3 adducts at approximately 26 min (11). The much earlier-eluting peaks observed from tobacco smoke adducts may indicate that the trapped radicals are quite polar, i.e., contain significant numbers of hydroxyl (-OH), amino (-NH2), or nitro (-NO2) groups. As discussed below, the presence of nitro groups in these species is predicted by mechanisms of radical formation through addition of NO2 to alkenes.
The prime suspect in postcombustion processes leading to radical generation in smoke plumes is NO2, which is produced by the slow reaction of NO and O2 (Reaction Scheme 1) (1,2,10).
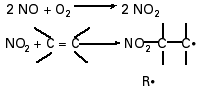
This reaction occurs with a third-order rate constant of 1.45 104/M2/sec at 298K. Because of its second-order dependence on NO, this reaction is unimportant in the atmosphere; however, in smoke, where NO concentrations are considerably higher, the conversion can occur at an appreciable rate. Reaction Scheme 1 illustrates how NO2 can react with unsaturated hydrocarbons to produce radicals minutes after combustion. Rate constants are available for a number of such reactions, e.g., k = 1.08 102/M/sec for NO2 + isoprene, at 298K (15). Oxygen-centered radicals such as OH, OR, OOR, and OOH may also be key participants in chain reactions within smoke [Reaction Scheme 2 (15)].
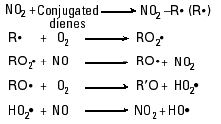
The presence of NO is again critical for those reactions because it provides a mechanism for conversion of peroxy radicals (OOR and OOH) into the more reactive alkoxy (OR) and hydroxy (OH) compounds, while simultaneously regenerating NO2. The generation of OH and OR can contribute to the production of additional carbon-centered radicals through radical addition and H-abstraction reactions (Reaction Scheme 3).
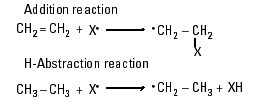
An additional possible source of NO2 in downstream smoke is decomposition of meta-stable compounds such as peroxyacetyl nitrate, a species currently implicated as an NOx reservoir in the atmosphere (16-18).
Any of the above processes or a combination of them could account for the arrays of radicals detected by our HPLC method. Identification of the radical adduct responsible for each HPLC peak would further elucidate the mechanism and smoke compounds involved, and efforts are underway to identify the trapped species via HPLC-MS techniques.
In the absence of detailed information about the adducts, we employed model systems to show that carbon-centered radicals can be formed and trapped from gas streams containing NO and an alkene. We selected two model systems, consisting of NO, air, and isoprene, with or without methanol, based on work done by others (2-5,10). We chose these compounds because of their relatively high abundance in cigarette smoke and their potential for radical production.
Figure 5 shows the carbon-centered radical suites collected from the two model systems of isoprene and NO both with and without methanol. The two chromatograms are nearly identical; both models produce a similar pattern of major peaks around 13, 15, 17, and 20 min, and a series of smaller peaks eluting both earlier and later. The retention times of the peaks at 3-5 min were not reliably reproducible and may represent an artifact. However, because these peaks did not appear in the blanks, they were included in the integrations; they contributed less than 5% of the total peak area. The appearance of four major peaks in the chromatogram is notable: Isoprene has two double bonds each of which could be subject to addition at either end to give four possible NO2 addition products. Two additional products are predicted from resonance structures of initially formed radicals. The peaks would be expected to have different intensities, controlled by the relative rates of reaction at each position and trapping of the resulting radicals. Additional peaks are possible from resonance structures or reactions with oxygen radicals as described above.
An interesting change in the chromatogram resulted from the addition of methanol to the gas stream: A small shoulder peak at 14 min was eliminated. Methanol is an excellent scavenger for OH (producing formaldehyde); thus the 14-min peak may represent a radical formed through hydroxyl reactions that were suppressed by methanol. The fact that addition of methanol did not significantly decrease the total population of trapped radicals indicates that hydroxyl radicals did not contribute substantially to the concentration of carbon-centered radicals in the isoprene-NO2 reaction. Methanol is most likely to influence the production of alkoxy radicals, which are not trapped by nitroxides and hence would go unnoticed here.
Interestingly, the peaks at 13 and 15 min produced from the model systems correspond to peaks of the same retention time found in the tobacco smoke radical chromatograms. Although the peak heights and ratios are not identical, the retention times are similar. However, the model systems failed to produce peaks at 7, 9, and 10 min, where all three tobacco samples had significant peaks. The later eluting peaks in the model systems, at 16 and 20 min, also do not correspond well to any in tobacco smoke. It would, of course, have been remarkable had isoprene and NO models reproduced the complete radical suites encountered in the vastly complex mixtures of tobacco smoke. But it is highly encouraging that a) isoprene-air-NO mixtures produce radicals that are similar in character to those formed in smoke, based on elution times of adducts; b) carbon-centered radicals were formed in room-temperature gases on time scales similar to those observed for tobacco smoke; and c) even a system with only one organic compound, isoprene, can produce a large number of radical products from reactions with NO and oxygen.
The interactions of NO, NO2, and alkenes in cigarette smoke have been supported by experimental results of Cueto et al (5,14) that demonstrated the more rapid loss of NO and concomitant rise in NO2 over about 800 sec in the presence of conjugated alkenes in air versus just NO and air. In those studies, FT-IR was used to monitor NO and NO2 concentrations in cigarette smoke and in model systems containing NO, air, and hydrocarbons. Upon addition of isoprene and/or methanol into those model systems, NO decreased and NO2 increased at rates more consistent with those observed in cigarette smoke samples (5). Although our model testing system was quite different than the one used by Cueto et al. (5,12) (Cueto used higher gas flow rates and higher initial isoprene and NO concentrations, and monitored NO/NO2 instead of R), both systems are consistent with the proposed mechanisms.
The delayed generation of radicals was also observable in our model system, as demonstrated in Figure 6. With the given concentrations of NO and isoprene, the maximum number of radicals was observed after about 20 sec of reaction. Once these radicals are formed they have very short lifetimes, so changes in the number of radicals trapped indicate their formation rate at the time the smoke passed through the trapping column. The rate-limiting step for radical formation is undoubtedly the production of NO2 from NO. Thus, the peak in radical production at 20 sec represents a peak in NO2 concentration; at longer times NO2 has been depleted by reaction with isoprene or other processes. Because the NO2 production reaction is second order in NO, the timing of the maximum radical formation rate is expected to be quite sensitive to the NO concentration. However, NO and/or NO2 were not rapidly depleted, as evidenced by the fact that measurable radicals were still detectable after smoke had aged for nearly 2 min.
In Table 2 we list the numbers of radicals trapped from each system investigated. The three types of tobacco show a large range, with a cigar producing nearly 3.5 times more trapped radicals than a cigarette. Model systems had lower levels of radicals trapped per volume sampled, as might be expected from a simple gas mixture. Both numbers and types of organic constituents in the gas are expected to influence the amount of trapped R. Perhaps most important, the concentration of NO in the gas stream, by controlling the rate of NO2 production, can determine when and where R are formed. The numbers in Table 2 represent integrations over the time the smoke or gas was in contact with the trap-coated surface. As illustrated by Figure 6, the number of trapped radicals varied as the gas mixture aged.
An equally critical issue is the competition among several pathways for reaction of R. Competitive reactions make it impossible to trap all radicals from a smoke plume, but a simple kinetic analysis allows us to estimate the relative importance of various fates of R. In a smoke plume, radicals may react with oxygen, a myriad of organic compounds, other radicals, or perhaps airborne particles. An additional pathway is provided in our trapping column: reaction with 3AP. The probability that R will be lost by a given route is determined by the rate constant for the reaction and the concentration of the reactants. In smoke, the vast majority of carbon-centered radicals are consumed by oxygen because O2 has both a large rate constant for reaction with R and a concentration orders of magnitude higher than that of any other reactive species in the system. (The large air input during smoking provides oxygen levels in smoke very near the atmospheric value of 20% v/v.)
With the trapping column in place, the number of radicals captured depends on the rate constant for the 3AP + R reaction and the effective concentration of 3AP, where the effective concentration is given by total surface area of the beads in the column and their coverage (3AP/area). That is, the higher the trapping surface area, the more R will be trapped before reacting with O2.
Because we know, or can estimate, rate constants and concentrations for several critical compounds, we can calculate what fraction of the total R present is trapped by 3AP in our system. Equation 1 describes the kinetics of these heterogeneous reactions and is used for comparing relative reaction rates for the competing reactions.
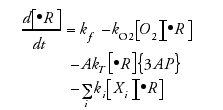 [1]
[1]
kf is the formation rate of radical R; kO2 is the second order rate constant for reaction of R with O2; [O2], [R], and [Xi] are concentrations of the respective species; kT is the rate constant for reaction of R (gas-phase) with adsorbed 3AP; A is the specific surface area of the trapping surface (area of adsorption/reaction volume); {3AP} is the surface concentration of 3AP (molecules/square centimeters); and ki are rate constants for other unspecified radical scavengers, Xi. Multiple radical species have been combined and designated collectively as R for simplicity.
Carbon-centered radicals react at near diffusion-limited rates with oxygen, for example kO2 = 6.6 108/M/sec (1.1 10-12 cm3/mol/sec) for the gas-phase reaction of CH3 with O2 (19). Solution bimolecular rate constants for reactions of nitroxides with carbon-centered radicals are 1.2 109/M/sec (2.0 10-12 cm3/mol/sec) (20); this value was taken to represent the surface phase reaction, because no literature data for that system were available. Although there are other species (Xi = alkenes, alkanes, NO, and so on) available for reaction with R, to a first approximation they can be neglected because of their low concentrations relative to O2 and 3AP. The most likely competitive scavengers for R in smoke--methanol and isoprene--have concentrations that are about half that of 3AP in the trapping system. Assuming pseudo-first-order reactions with O2 and 3AP, because they are in great excess, the pseudo-first-order rate constants are kO2[O2] and kTA{3AP}, denoted k´O2 and k´T, respectively. Using values of [O2]= 4.92 1018 mol/cm3 (20% v/v), A = 150 cm2/10cm3, {3AP} = 1.5 1015 mol/cm2, and the rate constants given above, we calculate pseudo-first-order rate constants of k´O2 = 5.4 106/sec and k´T = 4.5 104/sec. The fact that k´O2 is more than two orders of magnitude greater than k´T indicates that approximately 100 times more radicals react with oxygen than are trapped by 3AP in our current configuration.
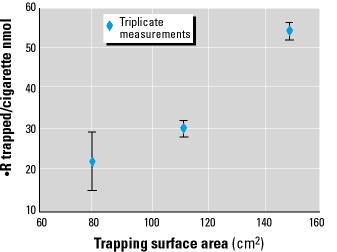
Either decreasing the O2 or increasing the 3AP concentration would enhance the trapping efficiency for radicals. The oxygen concentration cannot be decreased conveniently without changing the combustion process. Thus, competitively trapping radicals with 3AP requires changing A or {3AP}. It is not possible to increase {3AP}, because the bead surface already has 100% coverage; once a layer of 3AP is adsorbed to the glass beads, addition of more compound adds layers without increasing the amount available for reaction. The only remaining method to increase trapping efficiency is to increase A, the surface area of exposed 3AP. The success of this approach is shown in Figure 7 for three different values of A. Increasing the surface area of exposed 3AP clearly led to an increase in the number of radicals trapped. The expected behavior of this curve is a steady increase in trapped R with increasing 3AP until its concentration is high enough to trap essentially all radicals present, after which it levels off at a maximum value; i.e., additional increases in 3AP do not lead to trapping of additional radicals (13). Unfortunately, it is difficult to increase A sufficiently (a factor of 1,000) to out-compete oxygen, so we do not attain the maximum value. However, it is not necessary to trap every R to understand this type of system; it is sufficient to identify the important players and determine their concentrations and rate constants as outlined above.
|
Figure 7. Dependence of the number of radicals trapped from the gas-phase on the amount of surface area available for 3AP loading. Each data point represents triplicate measurements.
|
The kinetic analysis helps explain the variation in reported radical populations in tobacco smoke. In our preliminary study (11) using 3AP to trap radicals directly from the gas-phase, we reported trapping 22 ± 7 nmols R per Marlboro cigarette, compared with 54 ± 7 nmol in the current study; this change is a result of increasing the trap surface area from 79 cm2 to 149 cm2 by decreasing the bead size from 6 mm to 3 mm. The actual radical concentration is closer to 5,000 nmol/cigarette according to the kinetic analysis, which indicates that only about 1% of the radicals present were measured.
Several researchers have studied cigarette smoke radicals by bubbling smoke through a solution of spin trap (1,2,4,10) or blowing it over silica gel coated with spin trap (4,10), to collect both carbon- and oxygen-centered radicals, followed by detection with EPR. Using solution-phase spin trapping, Pryor et al. (2) reported 5 nmol gas-phase R per 1R1 research cigarette. Solution bimolecular rate constants for the reaction of radicals with spin traps (106-107/M/sec) (12) are approximately an order of magnitude lower than those of nitroxides (108-109/M/sec). This difference may explain the approximately 10-fold difference between the number of carbon-centered radicals trapped in Pryor and colleagues' reports compared with our experiments. However, because of uncertainties in relative trapping efficiencies and competitive reactions, as well as the different tobacco types or smoking methods employed by Pryor et al. (2), it is not productive to directly compare the numbers in greater detail.
In inhaled smoke, radicals are confronted with an environment similar to that of our trapping column. The lungs present a very large surface area, presumably coated with compounds that could react with carbon-centered radicals. With information about the concentrations of such compounds, an identical kinetic analysis could be performed to estimate the number of R that react with lung tissues compared with the number that react with oxygen. (Of course, those radicals that react with oxygen may go on to become equally damaging alkoxy or peroxy radicals, although perhaps attacking tissues via different pathways.)
Many carbon-centered radicals are present in aging cigarette smoke. With the described solid/gas-phase detection technique, each tobacco type tested produced a distinctly different suite of carbon-centered radicals representing a "fingerprint" of the source. The ability of this method to identify specific radical fingerprints for different gas-phase sources makes it unique to standard spin-trapping methods. Because it can rapidly and simultaneously detect multiple carbon-radical species, it provides a sensitive means to screen for a variety of potentially harmful radical species. If specific radicals were identified as having more or less potential to cause biologic damage, this technique could be used to identify which systems present the greatest risks.
Model systems consisting of NO, isoprene, and air, with or without methanol, produced chromatograms with 4 major and several minor peaks representing carbon radicals derived from isoprene. Several of these HPLC peaks coincided with those observed in tobacco smoke samples, but confirmation of radical identities will have to await HPLC-MS studies. The model systems also demonstrated the delayed production of radicals, supporting the NO/NO2/alkene reaction mechanism.
A kinetic analysis, supported by trapping experiments with varying trap surface area, shows a considerable competition for the carbon radicals between the 3AP radical scavenger and oxygen. About 40-75 nmol carbon radicals were trapped per liter of tobacco smoke, but approximately 100 times this many are calculated to have reacted with oxygen rather than the 3AP trap. This competition has direct parallels to processes in the lungs. The overall sequence may be described as follows: Inhaled smoke produces NO2 with a rate dependent on its NO concentration. NO2 then attacks alkenes to generate carbon-centered radicals. These radicals may react either directly with lung tissues or with oxygen to be thereby converted to oxygen radicals. The relative numbers of carbon radicals that react by each pathway will be determined primarily by the available surface area of radical-reactive species.