
| | | | |
Research
|
| Arsenic as an Endocrine Disruptor: Arsenic
Disrupts Retinoic Acid Receptor– and Thyroid Hormone
Receptor–Mediated Gene Regulation and Thyroid
Hormone–Mediated Amphibian Tail Metamorphosis Jennifer C. Davey, Athena P. Nomikos,
Manida Wungjiranirun, Jenna R. Sherman, Liam Ingram, Cavus
Batki, Jean P. Lariviere, and Joshua W. Hamilton Department of Pharmacology & Toxicology,
and Center for Environmental Health Sciences, Dartmouth Medical
School, Hanover,
New Hampshire, USA Abstract
Background: Chronic exposure to excess arsenic in drinking water has been strongly associated with increased risks of multiple cancers, diabetes, heart disease, and reproductive and developmental problems in humans. We previously demonstrated that As, a potent endocrine disruptor at low, environmentally relevant levels, alters steroid signaling at the level of receptor-mediated gene regulation for all five steroid receptors. Objectives: The goal of this study was to determine whether As can also disrupt gene regulation via the retinoic acid (RA) receptor (RAR) and/or the thyroid hormone (TH) receptor (TR) and whether these effects are similar to previously observed effects on steroid regulation. Methods and results: Human embryonic NT2 or rat pituitary GH3 cells were treated with 0.01–5 µM sodium arsenite for 24 hr, with or without RA or TH, respectively, to examine effects of As on receptor-mediated gene transcription. At low, noncytotoxic doses, As significantly altered RAR-dependent gene transcription of a transfected RAR response element–luciferase construct and the native RA-inducible cytochrome P450 CYP26A gene in NT2 cells. Likewise, low-dose As significantly altered expression of a transfected TR response element–luciferase construct and the endogenous TR-regulated type I deiodinase (DIO1) gene in a similar manner in GH3 cells. An amphibian ex vivo tail metamorphosis assay was used to examine whether endocrine disruption by low-dose As could have specific pathophysiologic consequences, because tail metamorphosis is tightly controlled by TH through TR. TH-dependent tail shrinkage was inhibited in a dose-dependent manner by 0.1– 4.0 µM As. Conclusions: As had similar effects on RAR- and TR-mediated gene regulation as those previously observed for the steroid receptors, suggesting a common mechanism or action. Arsenic also profoundly affected a TR-dependent developmental process in a model animal system at very low concentrations. Because RAR and TH are critical for both normal human development and adult function and their dysregulation is associated with many disease processes, disruption of these hormone receptor–dependent processes by As is also potentially relevant to human developmental problems and disease risk. Key words: arsenic (As) , CYP26A, deiodinase (DIO1) , endocrine, retinoic acid (RA) , steroid, thyroid (TH) . Environ Health Perspect 116: 165–172 (2008) . doi:10.1289/ehp.10131 available via http://dx.doi.org/ [Online 26 October 2007]
Address correspondence to J.W. Hamilton, Department of Pharmacology & Toxicology, 7650 Remsen Building, Room 514, Dartmouth Medical School, Hanover NH 03755-3835 USA. Telephone: (603) 650-1316. Fax: (603) 650-1129. E-mail: josh.hamilton@dartmouth.edu Supplemental Material is available online at http://www.ehponline.org/members/2007/10131/suppl.pdf We thank J. Bodwell, J. Gosse, and B. Stanton for their useful comments and suggestions. We also acknowledge the assistance of B. Jackson and A. LaCroix-Fralish of the Dartmouth Trace Elements Analysis (TEA) Core Facility for their assistance in the trace analysis of our samples. This work was supported by grant P42 ES07373 to J.W.H. [Superfund Basic Research Program (SBRP) Project, Project 2 ; National Institute of Environmental Health Sciences, National Institutes of Health]. A.P.N. was supported by a graduate fellowship (P42 ES07373 ; SBRP, Training Core) . The TEA Core is partially supported by grant P42 ES07373 (SBRP, Core B) and by an instrument grant from the National Science Foundation (MRI-0215913) . The authors declare they have no competing financial interests. Received 1 February 2007 ; accepted 25 October 2007. |
|
|
|
Exposure to excess arsenic,
principally from contaminated drinking water, is considered one
of the top
environmental health threats both in the United States and
worldwide [Abernathy et al. 2003; Mukherjee et al. 2006;
National Research Council (NRC) 1999, 2001; Smith et al. 2002;
Watanabe et al. 2003]. The majority of this exposure is from
natural geological sources of As that contaminate groundwater.
Epidemiologic studies have linked chronic exposure to
drinking-water As with increased risks of various cancers,
including those of the lung, bladder, skin, and liver, as well
as numerous other noncancer illnesses including vascular and
cardiovascular disease, diabetes, developmental and
reproductive problems, and neurologic and cognitive problems
(Abernathy et al. 2003; NRC 1999, 2001; Smith et al. 2002;
Tapio and Grosche 2006; Wasserman et al. 2004; Watanabe et al.
2003). Public water supplies in the United States and Europe
currently have a regulatory limit of 10 ppb As (0.13 µM).
However, a large segment of the population in the United States
and worldwide obtains its drinking water from private
unregulated wells. Also, many other areas of the world have
higher regulatory limits or water supplies that are
unregulated. Thus, As contamination continues to be a serious,
ongoing public environmental health problem affecting hundreds
of millions of people.
There are a number of proposed
mechanisms for the ability of As to influence so many diverse
disease
processes. These include alterations in cell signaling, cell
cycle control, oxidative stress, DNA repair, and others
(Abernathy et al. 1999; Aposhian and Aposhian 2006; Kitchin
2001; Rossman 2003). Moreover, it is becoming clear that there
are important dose-, time-, and tissue-specific differences in
effects of As, as well as important gene–environment and
co-exposure interactions that complicate how As alters disease
risk under any particular exposure circumstance (Andrew et al.
2003, 2006; Bodwell et al. 2004, 2006; Karagas et al. 2004;
Waalkes et al. 2003). We previously reported that As is a
potent endocrine disruptor, altering steroid hormone receptor
(SR)-mediated gene regulation at very low, environmentally
relevant concentrations in cell culture and in whole-animal
models (Bodwell et al. 2004, 2006; Davey et al. 2007;
Kaltreider et al. 2001). We have demonstrated that all five
steroid receptors (SRs) [i.e., the receptors for glucocorticoid
(GR), androgen (AR), progesterone (PR), mineralocorticoid (MR),
and estrogen (ER) hormones] are affected in a similar manner,
suggesting a broad effect on these pathways and also suggesting
a common mechanism for these effects (Bodwell et al. 2004,
2006; Davey et al. 2007; Kaltreider et al. 2001). Given this
strong disruption on this entire class of nuclear hormone
receptors, we were interested in whether these effects extend
to other members of the larger nuclear hormone receptor
superfamily.
In this study we
examined the effects of As on gene regulation by two related
class II receptors, the
retinoic acid (RA) receptor (RAR) and the thyroid hormone (TH)
receptor (TR). Both receptors normally partner with the
retinoid X receptor (RXR) to form heterodimers that act as
transcription factors, binding to the RA-response element
(RARE) of RA-inducible genes or the TH-responsive element (TRE)
of TH-inducible genes, respectively (Mark et al. 2006; Tsai and
O’Malley 1994; Wong and Shi 1995; Zhang and Lazar 2000).
We report here that As has significant effects on both RAR- and
TR-mediated gene regulation at very low doses (0.01–2 µM),
and that these effects are strikingly similar to those
previously observed with the SRs, suggesting a common
mechanism. We also investigated whether such As-induced
alterations in hormone signaling would lead to pathophysiologic
consequences, using amphibian tail metamorphosis as a model
system. Anuran metamorphosis is highly dependent on TH- and
TR-mediated processes (Buchholz et al. 2006; Buckbinder and
Brown 1993; Wong and Shi 1995; Yaoita and Brown 1990) and is
known to be perturbed by agents that interfere with TH or TR
signaling (Buchholz et al. 2003; Lim et al. 2002). Because TH
is also important for many aspects of mammalian embryonic development
(Galton 2005; Oppenheimer and Samuels 1983; Porterfield and
Hendrich 1993), this is a useful model for understanding
possible effects on human development. Arsenic had profound
effects on TH-mediated ex vivo tail metamorphosis at very low,
environmentally relevant concentrations, suggesting that there
are likely to be
other developmental and pathophysiologic effects of low dose
As endocrine disruption in vivo.
NT2 cell culture and transfections. NT2
[NTERA-2; American Type Culture Collection (ATCC), Manassas, VA]
human embryonic carcinoma cells were
maintained in Dulbecco’s Modification of Eagle’s
Medium (DMEM; Invitrogen, Carlsbad, CA) plus 10% fetal bovine
serum (FBS; Atlanta Biologicals, Norcross, GA). Cells were
split into 6-well plates at 1.5 x 105 cells/well and
grown in phenol red–free
DMEM plus 10% charcoal-stripped FBS overnight for transfection
experiments. The construct used was generously provided by
James DiRenzo (Dartmouth Medical School). Briefly, the
construct contains two canonical tandem RARE sequences
(5´-AGGTCA-(N)5-AGGTCA-3´) upstream of a thymidine kinase
promoter and the firefly luciferase coding region with a pBR322
backbone (RARE-LUC) (Kurokawa et al. 1994). Cells were
transfected with 250 ng of the RARE-LUC construct using Fugene
(Invitrogen) according to the manufacturer’s recommended
protocol. All-trans-retinoic acid (ATRA; Sigma Chemical Co.,
St. Louis, MO), reconstituted in DMSO and stored at
–20°C, was used as ligand at concentrations
indicated.Arsenic (As, As+3, sodium arsenite, NaAsO2;
Sigma Chemical Co.) was dissolved in water and kept frozen until
the
day of use. Treatments were performed 24 hr after transfection.
ATRA and As were added simultaneously at the doses and for the
durations described in "Results."
|
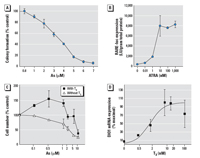
Figure 1. Dose–response
curves for As, ATRA, and TH in NT2 and GH3 cells calculated
using average values for each dose. Data points represent the
mean ± SE of data from three separate experiments. (A) Cytotoxicity of As
in NT2 cells exposed to As for 24 hr and assessed by
colony-forming assay. Data are expressed as colony formation as
a percent of the control. (B) Induction of RARE-luc expression
in NT2 cells by ATRA. RARE-luc expression is expressed as mean ± SE
luciferase (LU) per gram protein. The median effective
concentration (EC50) for ATRA induction is approximately 7 nM. (C) Cytotoxicity of As
in GH3 cells assessed essentially as described for (A), except that cells
were cultured in media plus stripped serum with or without 10 nM
T3. (D) Induction of DIO1 expression by T3 in GH3 cells assessed essentially as described
in (C),
except that DIO1 mRNA was assessed by RT-PCR. Data are
expressed as a percent of the maximum value. See "Methods" for
details.
|
|

Figure 2. Effects
of As on ATRA induction of RARE-luc expression in NT2 cells.
Cells were transfected with the RARE-luc construct 24 hr before
treatment with 10 nM ATRA with or without simultaneous
treatment with As for 24 hr. See "Methods" for
details. Data are expressed as mean ± SE of the values
from replicates of experiments. Bars that do not have the same
letter are significantly different from each other at p < 0.003
using an unpaired t-test.
|
|
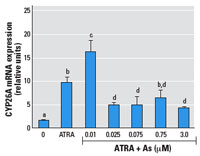
Figure 3. Effects
of As on ATRA induction of CYP26A mRNA expression in NT2 cells.
Cells were treated with 10 nM ATRA with or without
simultaneous addition of As for 24 hr; mRNA expression was
measured by RT-PCR. See "Methods" for details. Data
are expressed as mean ± SE of the values from replicates
of experiments. Bars that do not have the same letter are
significantly different from each other at p < 0.003
using an unpaired t-test.
|
|
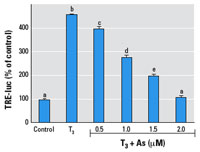
Figure 4. Effects of As on T3 induction of TRE-luc expression in GH3 cells.
Cells were transfected with the TRE-luc construct 24 hr before
treatment with 2 nM T3 with or without simultaneous
treatment with As for 24 hr. See "Methods" for details. Data are
expressed as mean ± SE of the values from replicates of
experiments. Bars that do not have the same letter are
significantly different from each other at p < 0.003
using an unpaired t-test.
|
|

Figure 5. Effects of As on T3 induction of DIO1 mRNA expression in GH3 cells.
Cells were treated with 2 nM T3 with or without As, and DIO1 mRNA expression was
measured 6 hr (A) or 24 hr (B) after treatment.
See "Methods" for
details. Data are expressed as mean ± SE of the values
from replicates of experiments. Bars that do not have the same
letter are significantly different from each other at p < 0.01 using
pairwise Student’s t-test analysis.
|
|
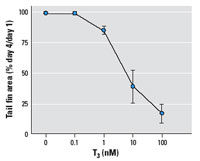
Figure 6. Effects
of T3 on tail fin shrinkage of Xenopus tadpole tails cultured ex vivo as described in
"Methods." Data are expressed as mean ± SE
of values from six tails per treatment in three separate
experiments.
|
|

Figure 7. Effects
of As on T3-mediated tail shrinkage in Xenopus tadpole tails
cultured ex vivo shown by representive samples from tail
resorption experiments. See "Methods" for details.
Morphometric software was used to trace the tail fin area
(shown in black), which was used to calculate the differences
in area for each tail between day 1 and day 4. Results from
these experiments are shown in Figures 6 and 8.
|
|
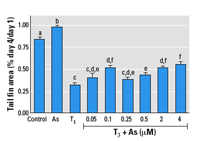
Figure 8. Effects of As on T3-mediated tail shrinkage in Xenopus tadpole tails
cultured ex vivo. See "Methods" for details. Data are
expressed as mean + SE of values from 5–6 individual
tails per experiment and 4–8 individual experiments per
treatment. Bars that do not have the same letter are
significantly different from each other at p < 0.01 using pairwise
Student’s t-test analysis.
|
Clonogenic cell survival assay for NT2
cells. NT2
cells were grown in phenol red–free DMEM media plus 10% FBS and split into 6-well
plates with approximately 100 cells/well and allowed to grow
overnight. Cells were then exposed to 1.0–7.0 µM
As plus 10 nM ATRA for 24 hr. Next, fresh media without As but
including 10 nM ATRA was added, and cell growth was checked
daily. When colonies of ≥ 25 cells were formed in control wells,
the media was removed and the cells were fixed in 1% formaldehyde
for 1 hr and then exposed to 100% methanol for 5 min. Methanol
was removed and cells were stained with 0.3% Giemsa stain (in
70% methanol) for 15 min. Colonies were rinsed twice with
phosphate-buffered saline (PBS) and counted. Results are given
as a percent of control, and each condition was repeated in
triplicate.
GH3 cell culture and transfections. GH3 rat pituitary
tumor cells (ATCC) were cultured in F-12 Nutrient Mixture (Ham)
(Invitrogen) supplemented with 15% horse serum (Atlanta
Biologicals) and 2.5% FBS. For triiodothyronine (T3)/As
treatments, 6-well plates were seeded at 350,000–750,000 cells/well
and cultured in F-12 medium for 3 days. Cells were then put
into phenol red–free DMEM supplemented with 10%
charcoal-stripped FBS for 18 hr prior to treatments. The TR
response element–luciferase (TRE-luc) construct used for
transfections was created by exchanging the glucocorticoid
response element (GRE) sequence with two canonical TRE direct
repeats in the pXP2-GRE–luciferase construct kindly
contributed by J. Bodwell (Dartmouth Medical School). The final
construct was sequenced to verify successful insertion.
Transfections were performed with Lipofectamine Plus
(Invitrogen) with the following optimizations. Twenty-four
hours after plating, media was changed to unsupplemented
Opti-MEM (Invitrogen), then transfection followed using the
manufacturer’s protocol. Three hours later, Opti-MEM was
replaced with phenol red–free DMEM plus 10%
charcoal-stripped FBS for the duration of the experiment. Cells
were treated withAs as indicated. T3 (CAS 55-06-1;
Sigma) was reconstituted to 1 mM in 45% propylene glycol (1,2-propanediol;
Aldrich, St. Louis,
MO), 45% water, and 10% 0.1 N sodium hydroxide and stored at
4°C covered.
Clonogenic cell survival assay for GH3
cells. Clonogenic
assays were performed by plating approximately 5,000 cells/well
in a 6-well
plate and allowing them to grow in F-12 Nutrient Mixture for
a
total of 3–4 days. After an 18-hr treatment in phenol
red–free DMEM with 10% charcoal-stripped serum, cells
were treated for 24 hr with various doses of As, with or
without T3. Arsenic was removed and the cells were
allowed to grow for approximately 5 days in phenol red–free
DMEM plus 10% charcoal-stripped serum with or without T3. Cells
were trypsinized, resuspended in PBS, and counted in azide-free
isotonic diluent (Valtech Diagnostics, Inc., Brackenridge, PA)
using a Coulter Counter Z1 (Beckman Coulter, Fullerton, CA).
Cells were counted individually rather than via colony
formation (as with the NT2 cells) because GH3 cells would not
adhere under the conditions necessary for staining the
colonies.
ICP-MS analysis of total arsenic in cell
culture media. Total
arsenic levels in cell culture media were measured by the Dartmouth
Superfund
Trace Metal Core Facility using collision cell inductively
coupled plasma mass spectroscopy (ICP-MS; Agilent
octopole/reaction cell 7500c ICP-MS; Agilent, Palo Alto, CA),
employing helium as the collision gas. The media was diluted
and analyzed by the method of standard additions. Method
detection limits were 0.005 µM, and the overall method
uncertainty was 8%. All media used contained undetectable
levels of As or levels well below any of the exogenously added
effective doses (i.e., < 0.01 µM).
Luciferase and protein assays. Cells
were rinsed twice with cold PBS, covered with 150 µl of Promega lysis buffer, and scraped from the
plate. Cell lysates were frozen at –80°C, thawed,
vortexed for 30 sec, and spun at 12,000 x g for
5 min; supernatants were then transferred to a new tube and stored
at –80°C. Each experimental condition in each experiment
was done in replicates of six. Transfection efficiency was
consistent, with little variability among the biological
repeats in each treatment group and low standard deviations
between 3 to 4 replicate experiments. We used Prism software
(GraphPad Software Inc., San Diego, CA) for all statistical
analyses. Luciferase assays were performed with the Promega
Luciferase Assay System kit (Promega, Madison, WI) following
the manufacturer’s recommended protocol. We used a
luminometer (Dynatech Laboratories, Chantilly, VA) to determine
the light units per sample. Protein assays were performed with
the Pierce BCA Protein Assay Reagent Kit (Pierce, Rockford, IL)
following manufacturer’s recommended protocol to
normalize the total level of protein in samples. Results of
protein assays were determined using a Thermomax microplate
reader (Molecular Devices, Sunnyvale, CA).
Semiquantitative real-time polymerase
chain reaction (PCR) for CYP26A or DIO1. We isolated
total RNA from the cells after indicated treatment times using
the Qiashredder and RNeasy Mini Kits
(Qiagen, Valencia, CA) or Trizol (Invitrogen) according to the
manufacturer’s recommended protocols. Contaminating
genomic DNA was removed using DNA-free kits (Ambion, Austin,
TX), and total RNA was quantified with a NanoDrop ND-1000
Spectrophotometer (NanoDrop Technologies, Rockland, DE). RNA
(1–2 µg) was reverse-transcribed (gene-specific
primer) with Omniscript Reverse Transcriptase (Qiagen), and
primers were synthesized to specifically amplify either the rat
type I deiodinase (DIO1) gene from the GH3 cells (GenBank accession
no.BC083557; National Library of Medicine 2007) or the human
cytochrome P450 26A (CYP26A) gene from the NT2 cells (GenBank
accession no. NM000783) [see Supplemental Material (http://www.ehponline.org/members/2007/10131/suppl.pdf) for details].
We performed semiquantitative real-time PCR (RT-PCR) to assess
relative quantities of each transcript with the 7500 Real-Time
PCR System (Applied Biosystems, Foster City, CA). Negative
controls, including samples that would either detect
contaminating genomic DNA in the RNA samples or contamination
of PCR reagents, were always included. On each plate we
included an internal standard curve for each transcript, which
consisted of a serial dilution of cDNA known to contain the
transcript in question. The curve generated from the plate was
then used to quantify the level of transcript for all the
experimental samples on the same plate. RT-PCR for 18S levels
was performed to normalize total RNA levels. Results were
unaltered by 18S normalization.
Xenopus
tadpole culture. We purchased Xenopus laevis tadpoles from
Nasco (Fort Atkinson, WI). Animals were treated humanely and with
regard to alleviation of suffering; all studies were
performed in compliance with institutional animal care and use
guidelines approved by Dartmouth Medical School (Protocol
04-03-11). Animals were acclimated to COMBO culture media (8.7
mg/L K2HPO4, 36.9 mg/L MgSO4, 62.8 mg/L NaNO3, 36.7 mg/L CaCl2, 28.42 mg/L Na2SiO3, 24 mg/L H3BO4, 12.6 mg/L NaHCO3
plus
2.18 µg/L H2SeO3,
16 µg/L
NaBr, 155 µg/L LiCl, 70 µg/L RbCl, 3.3 µg/L
KI, 150 µg/L SrCl2·6H2O,
pH 7.7) and carefully staged when they arrived. Animals were
kept in
COMBO media in 5-gal Nalgene buckets (Nalge Nunc International,
Rochester, NY) at room temperature until they reached stage 58
of development. Stage 58 was determined using staging criteria
determined by Nieuwkoop and Faber (1994). At stage 58, Xenopus
tadpoles have functional TR in tail tissue, and the tail will
metamorphose upon exposure to T3 whether or not it is attached to the animal
(Shi et al. 2002; Tata 1966).
Ex-vivo tail
culture. Tails
were excised from stage 58 tadpoles, dipped in 70% ethanol, and
immediately
placed in a 6-well plate with 3.0 mL phenol red–free
minimal essential media (MEM; Invitrogen) per well. Arsenic was
added to the media immediately (doses indicated in the
"Results"), and T3 was added to the media 18 hr later. Trial
experiments in which arsenic and T3 were added simultaneously
resulted in similar shrinkage patterns. Tail cultures were kept
in the dark at
approximately 20–23°C. Medium was changed daily, and
photographs were taken with a digital Nikon Coolpix camera
(Nikon Inc., Melville, NY). Experiments lasted approximately
5 days. Fins of tails treated with T3 only generally resorbed significantly or
completely in 4 days. Digital photographs were analyzed with
NIH Image software (National Institutes of Health, Bethesda,
MD) to assess the area of tail fin each day. The area of the
dorsal tail fin on day 4 was divided by the area of the dorsal
tail fin on day 1 to determine the percent of resorption. Each
treatment group in each experiment consisted of 6 tails, and
the experiment was repeated four to eight times. Occasionally
tails would not resorb at all when given T3 alone,
indicating that they did not have sufficient TR to
metamorphose, most likely as a result of incorrect staging. In
these cases, all tails in the experiment that did not resorb at
all when given T3 were removed from analysis regardless
of treatment; 3%–10% of the 42 tails in an experiment were
removed and they were evenly distributed between the groups.
Statistical significance was determined with Prism software
using an unpaired t-test and a p-value < 0.01 for the tail
fin area, except when determining the optimal T3 dose
where the value shown is mean ± SE.
Effects of As on RAR-mediated
transcription. Ligand-activated RAR forms heterodimers
with RXR and binds RAREs in RA-responsive gene promoters, which
induces
transcription (Mark et al. 2006). Initial dose–response
experiments (Figure 1B) demonstrated that 10 nM ATRA was an
adequate and physiologically relevant dose of ligand that
induces a significant increase in expression of the RARE-luc
construct when transfected into these cells. Cytotoxicity,
measured by colony-forming assays, demonstrated an LC50 (concentration
lethal to 50%) of 3 µM for NT2 cells
(Figure 1A). Very low concentrations of As (0.05–0.25 µM)
enhanced RA-inducible RARE-luc expression compared with RA
alone (Figure 2). Conversely, a higher but still noncytotoxic
concentration of 2.0 µM As strongly repressed
RAR-mediated induction of the construct (as did the higher but
slightly cytotoxic dose of 5 µM). The CYP26A gene is
endogenously expressed in NT2 cells, and a RARE in its promoter
facilitates its induction by ATRA (Loudig et al. 2000). The
transcript level of the CYP26A gene was measured by RT-PCR in
NT2 cells after exposure to 10 nM ATRA plus As (Figure 3). Similar
to the
RARE-luc construct, hormone induction of CYP26A mRNA
expression was enhanced by 10 nM ATRA plus As at a very low dose
(0.01 µM),
and its induction was repressed in a dose-dependent manner by
As doses of ≥ 0.025 µM.
TH-mediated transcription. GH3 cells
are a rat pituitary tumor cell line that expresses functional TRs
and expresses genes directly
induced by TH. We performed a series of initial
dose–response experiments to determine an effective and
physiologic dose of TH (administered as T3)
and found that 1–2 nM T3 was sufficient to induce
transcription (Figure 1D) of the DIO1 gene approximately 4-fold.
This was also sufficient to induce the TRE-luc construct 4-fold.
GH3 cells, like the NT2
cells, were moderately sensitive to the toxic effects of As,
with an LC50 of 5–10 µM, as determined by a
clonogenic cell survival assay (Figure 1C). Interestingly, the
clonogenic assay results indicated an increase in proliferation
rather than cytotoxicity when 0.1–1 µM As was
combined with T3 (the proliferative effect was removed when T3 was
removed). The LC50 was decreased to 0.9–2.0 µM
if the cells were grown in stripped serum without T3.
The TRE-luc construct was repressed in a dose-dependent manner
by
0.5–2.0 µM As (Figure 4). We then examined the
effects of As on TH induction of the endogenously expressed DIO1
(Jakobs et al. 1997) by measuring mRNA levels by RT-PCR 6 or
24 hr after
treatment. At 6 hr, concentrations of 0.01–2.0 µM
As produced a biphasic dose response similar to the NT2 results
with RA, with a significant super-induction at lower As
concentrations (0.01–0.1 µM) and repression at 2 µM
(Figure 5A). The response at 24 hr differed from the
RAR-mediated genes in that any repressive effects were gone,
and 1.0 and 2.0 µM As enhanced hormone-stimulated
expression. Arsenic doses > 2 µM produced significant
cytotoxicity, so we could not determine whether As suppresses
TH induction at 24 hr independent of general toxic mechanisms
that might compromise mRNA expression.
TH-directed amphibian tail metamorphosis. Based on the
effects of As on TR-mediated gene regulation described above,
we were interested in whether there were specific
pathophysiologic consequences of this endocrine disruption. We
assessed T3-dependent tail shrinkage by measuring the area
of the tail fin over a 4-day period, which is generally the
time it takes for the fin to completely resorb in our assay.
Fin resorption is the first step in visible tail metamorphosis,
is amenable to quantitation by morphometric analysis, and is
representative of the overall tail shrinkage process. A 10-nM
dose of T3 in the tail culture media was consistently
sufficient to induce tail fin resorption (Figure 6); thus, we
used this dose throughout the subsequent experiments. As shown
in Figure 7, treatment of tails ex
vivo with T3 led to
shrinkage of the tail fins that was readily apparent. Control
tail fins not exposed to T3 or As normally shrank
10–20% during an
experiment (Figures 7 and 8). Tails exposed to As alone shrank
slightly less than controls (Figure 8). In the As plus T3 groups,
the lowest As concentration used (0.05 µM) had little
effect compared with T3 alone, and the tail fins resorbed
approximately 58% in both groups (Figures 7 and 8). However,
concentrations
of As above this (0.5–4 µM) decreased T3-dependent
tail fin resorption in a dose-dependent manner (Figure 8). A
concentration of 0.25 µM As slowed resorption by 4%
compared with T3 alone, whereas 4 µM As decreased
resorption by 20%. Interestingly, the intermediate
concentration of 0.1 µM As did not follow the
dose-dependent pattern of the higher As doses; this finding is
discussed below. In summary, As significantly altered the T3-
and TR-dependent metamorphosis of Xenopus tails exvivo, indicating
that As disrupts T3 hormone
signaling through its receptor, TR/RXR, and has the potential
to affect hormone-
regulated embryonic development at low
physiologically and environmentally relevant concentrations invivo.
Based on our previous studies with SRs, we
were interested in whether As could also disrupt other, less
similar nuclear hormone receptors. Because we were interested
in examining the effects of As on hormone regulation of both
transiently transfected gene constructs as well as native
hormone-responsive genes, we chose two different cell culture
models, each optimal for the receptor system under study. Our
previous results (Davey et al. 2007), as well as those of
others (Kaltreider et al. 2001), strongly indicate that working
in the appropriate cell line is critical to mimicking the
effects of As on gene expression observed in vivo. Mann et al.
(2005) reported that As had no effect on ER-mediated gene
expression in heterologous systems such as COS1 cells, which
do not normally express ER but require co-transfection of an
ER-expressing plasmid. Lack of response in such systems, even
when the receptor is reexpressed, may indicate that other
critical aspects of the receptor machinery (e.g., key
coregulators that are critical for the As response) may be
missing. Also, the sensitivity of cultured cells to the
cytotoxic effects of As is highly cell line specific; thus, it
is important to know the dose response to As for a given cell
line so the appropriate dose range can be selected and the
toxic-equivalent doses between lines can be compared. In the
present study we found that NT2 cells are more sensitive to
the
toxic effects of As than other cell lines we have used. Also,
in order to examine the pathophysiologic consequences of
endocrine disruption of one of these type II receptors by As,
we chose another well-
characterized system, amphibian tail
metamorphosis, which is known to be highly TR-dependent (Brown
et al. 1996; Das et al. 2006; Dodd and Dodd 1976; Lim et al.
2002; Veldhoen and Helbing 2001).
We observed significant
effects of As at very low, noncytotoxic, environmentally relevant
concentrations
on both RAR- and TR-mediated gene expression in this study.
These As effects were very similar to those previously observed
for GR and the other SRs, suggesting a common mechanism of
action. As with SRs, the effects of As exhibited a complex
dose–response pattern that was biphasic, with As
significantly enhancing hormone-mediated gene expression at
very low doses while strongly suppressing gene expression at
higher, but still moderate, doses of As. Similar effects were
observed for GR, MR, PR, AR, and ER, although the precise
concentrations varied as a function of relative As toxicity in
each cell system (Bodwell et al. 2004, 2006; Davey et al.
2007). Our previous studies with SRs strongly suggest that the
receptors themselves are not the actual targets for these As
effects. First, the SRs share little absolute homology, yet
they respond almost identically to As (Bodwell et al. 2006;
Davey et al. 2007). Although GR, MR, AR, and PR share extensive
similarity, ER is much more distally related and shares little
absolute homology, but ER responds similarly to As (Davey et
al.
2007). Second, mutational studies with GR indicated that
neither the N-terminal regulatory or C-terminal ligand-binding
domains were necessary to elicit the effects of As (Bodwell et
al.
2004, 2006). Likewise, extensive mutational analysis of the
remaining central DNA-binding domain of GR failed to
demonstrate any critical region that could serve as the likely
target for As, nor is there sufficient homology among the SRs
to explain a common As effect. The present results show similar
effects of As on TR and RAR in spite of their even greater
divergence from the SRs; this further supports the hypothesis
that it is some common aspect of their regulation that is the
actual target.
The mechanism of gene activation
for TR and RAR differs from that of the SRs. Unlike the SRs,
which
form homodimers and bind to palindromic hormone-responsive
elements (HREs) in promoters, TR and RAR each normally form
obligate heterodimers with the RXR to form a functional
transcription factor. The TR-RXRs and RAR-RXRs are also
normally bound to their cognate response elements (TRE and
RARE, respectively) in hormone-responsive promoters in the
absence of ligand, unlike the SRs, which normally reside as
quiescent monomers in the cytoplasm (GR, MR, PR, AR) or nucleus
(ER) and then migrate to their respective HREs following
hormone activation and dimerization. Co-repressors normally
prevent transactivation of TR or RAR until ligand binding
occurs (Bastien and Rochette-Egly 2004; Hermanson et al. 2002;
McKenna and O’Malley 2002; Shi et al. 2002). Similar to
what was observed for SRs, As produced a biphasic response to
gene induction with RAR and TR. The suppressive effect of
intermediate doses of As is seen universally and strongly with
all of these nuclear hormone receptors under virtually all
experimental conditions, and it appears to be most closely tied
to transcription. In contrast, the low-dose As enhancement of
hormone induction is more variable among receptors, is more
susceptible to experimental manipulation, and appears to be
most closely associated with earlier steps of receptor
activation, based on detailed studies with GR (Bodwell et al.
2004, 2006). For example, certain GR mutants lack the low-dose
enhancement while retaining intermediate dose suppression by
As. Similarly, the low-dose enhancement can be progressively
dampened and eventually lost by progressively increasing the
number of hormone-activated receptors in the cell, although
higher dose suppression by As remains relatively constant
(Bodwell et al. 2004). We propose that the mechanism by which
As enhances hormone-induced gene activation at very low doses
is distinct, and can be separated experimentally, from the
suppressive effects seen at the slightly higher but still
noncytotoxic intermediate doses. However, the precise
mechanisms by which As elicits these two effects remains to be
determined.
RAR mediates RA signaling during embryonic
development. Either excessive or deficient levels of RA, a
derivative of vitamin A, results in teratogenic effects
(Shenefelt 1972; Thompson et al. 1964; Wolbach and Howe 1978).
RA signaling is also involved in tissue homeostasis, lipid
metabolism, and cellular differentiation and proliferation in
the adult (Shenefelt 1972). The family of retinaldehyde
dehydrogenases and Cyp26(A1,B1,C1) are RA-synthesizing and
RA-catabolizing enzymes, respectively. Transgenic mice without
CYP26A1 result in embryonic lethal phenotypes, thus indicating
a key role of CYP26A1 in
embryogenesis (Abu-Abed et al. 2001). The present study
indicates that RA-dependent CYP26A1 mRNA expression can be
enhanced or repressed by As exposure, depending on the dose of
As. CYP26A1 plays a key role in inactivating RA; therefore, its
dysregulation could have important pathophysiogic consequences,
including developmental processes, similar to those of the DIO1
gene (White et al. 1996, 1997). Because these effects were
observed
for
concentrations of As that are directly relevant to
environmental levels of As in drinking water of concern
throughout the United States and many other parts of the world,
this has potentially important implications for human
reproduction and development in exposed populations.
TR is the mediator of critical
TH-regulated processes in adults and during embryonic and fetal
development (Gothe et al. 1999). The active form of TH, T3, is
produced when type I or type II deiodinase removes a specific
outer ring iodine of thyroxine (T4). T3 is
the active TH ligand that binds TR, and therefore its production
is a crucial step in TH-driven gene
induction. The DIO1 gene has a TRE in its promoter and is directly
induced by T3 through TR. The change in expression
of DIO1 we
observed at 6 or 24 hr of As exposure indicates a transient superinduction
by As at very low doses and a transient repression by As at
higher doses. Differential effects of acute versus chronic As
exposure are of interest because TH levels—kept in
equilibrium by deiodinases and T3 levels in the pituitary—are
critical to thyroid function in the whole body. Thus, understanding
how As
alters T3 homeostasis will be an important aspect
of understanding the overall effects of As on this hormone
pathway, both for human development and in adult physiologic
processes.
The clonogenic assays with the GH3 cells
indicated enhancement of proliferation at low-dose As in the
presence of T3. T3 activates growth hormone in GH3 cells, causing
proliferation (Kitagawa et al. 1987; Kitamura et al. 2002), and
low-dose arsenic has also been shown to cause proliferation of
cells (Hwang et al. 2006; Vogt and Rossman 2001). Low-dose As
appears to enhance the proliferative effect of T3,
demonstrating another type of As-induced endocrine disruption
(i.e., enhancement of proliferation that is hormone dependent).
Metamorphosis of Xenopus laevis has been
extensively characterized (Buchholz et al. 2006; Buckbinder and
Brown 1993; Gudernatch 1912; Wong and Shi 1995; Yaoita and
Brown 1990). In vivo, the tadpole produces T3 in concert
with induction of TR, leading to TH-dependent alterations in
gene expression that regulate tail resorption (Brown et al.
1996; Das et al. 2006; Dodd and Dodd 1976). This can be
mimicked by culturing appropriately staged tadpole tails ex vivo
and exposing them to exogenous TH, leading to tail shrinkage
similar
to that
seen in vivo (Das et al. 2006; Lim et al. 2002; Tata 1966;
Veldhoen and Helbing 2001). Tadpole tails will respond to T3 in the
culture media essentially as they would as part of the whole
animal, by shrinking through what is thought to be a
combination of apoptosis and necrosis (Du Pasquier et al. 2006;
Nakajima and Yaoita 2003).
We assessed T3-dependent tail shrinkage
by measuring the area of the tail fin over a 4-day period, which
is generally the
time it takes for the fin to completely resorb in our assay.
Fin resorption is the first step in visible tail metamorphosis,
is amenable to quantitation by morphometric analysis, and is
representative of the overall tail shrinkage process. The ex
vivo tail culture
allows for the very controlled and T3-driven
metamorphosis of an entire tissue and, because the tadpole skin
is so permeable, both T3 and As are able to diffuse into the tissue from
the media (Lim et al. 2002; Veldhoen and Helbing 2001). Tail
resorption is completely dependent on T3 acting
as a ligand for TR and trans-activating genes necessary for
resorption, as shown in previous studies including those using
an antagonist to TR, which blocks these processes (Lim et al.
2002). The lowest As concentration that had an effect on tail
fin resorption (0.1 µM) did not follow the dose-dependent
pattern of all the higher As concentrations. (Figure 8). This
biphasic pattern was highly repeatable and statistically
significant, indicating two different dose responses over this
range and suggesting perhaps two different mechanisms of action
underlying these effects. Indeed, two different mechanisms of
interference could lead to the same overt results (i.e., less
fin shrinkage).
Tail resorption is only one piece of the
complete transformation of the aquatic tadpole into the
terrestrial frog (Das et al. 2006; Dodd and Dodd 1976). Tissues
in the animal are completely remodeled to function on land.
In
the space of a few weeks, body parts are generated (limbs)
while others completely resorb (tail); cell death, growth, and
differentiation can occur simultaneously in a single tissue
(intestine, eye, blood, skin). All this transformation is
directed by TH. If TH is removed, metamorphosis does not happen
and a continuously growing tadpole will result (Allen 1916;
Dodd and Dodd 1976). When TR is expressed and T3 is
produced, the program for the transcription of genes necessary
for metamorphosis in any tissue is ready to proceed. This model
has relevance to human development because the plasma levels of
TH spike in humans during the perinatal period (6 months of
gestation through several months postnatal), which correlates
temporally with the TH spike in amphibians during metamorphosis
(Shi et al. 2002). Extreme thyroid deficiency in humans at
birth leads to cretinism, with characteristic mental
retardation, short stature, and hearing loss. These severe
deficiencies are detected at birth and can be quickly treated
with exogenous hormone, but subtler effects may not be as
evident at birth (Galton 2005; Oppenheimer and Samuels 1983;
Raz and Kelley 1997). Indeed, subtle effects on cognitive
function have been noted in epidemiology studies of children
exposed to excess As in drinking water (Wasserman et al. 2004).
Studies showing disruption of thyroid hormone functions after
As exposure in rodents include an increase in thyroid cancer
(Yamamoto 1995), a synergistic effect between As and TH on
oxidative stress (Allen and Rana 2003), and a disregulation of
deiodinase levels in fetal brain with concurrent As exposure
and selenium depletion (Miyazaki et al. 2005).
In the present study we have demonstrated
that As can act as a potent endocrine disruptor not only for
the entire SR family but also for two important members of the
larger nuclear hormone receptor superfamily. It seems likely,
based on the ubiquity of these effects and their likely common
or shared mechanism(s), that As will also have similar effects
on other members of this large hormone receptor superfamily.
It
will be important to determine the extent to which this occurs
and under what conditions, but also to investigate the
biological consequences of such effects. Including the present
study, we have demonstrated in two different in vivo systems
that there are specific and predictable pathophysiologic effects
of
endocrine disruption by As. We previously reported that As has
profound effects on the GR-dependent freshwater-to-saltwater
transition of killifish (Fundulus
heteroclitus) (Shaw et al. 2007;
Stanton et al. 2006).
Exposure to excess As in drinking water
has been associated with an extensive and growing list of
disease risks. Given the important and myriad roles of hormones
and their receptors in normal physiology and in the
pathophysiology of these and other diseases, it is likely that
endocrine disruption by As plays an important role. Previously,
it was thought that because As does not significantly
accumulate in the body, unlike persistent organics or metals
such as mercury and lead, and also does not cause DNA damage
or
mutations, its effects on disease risk might also be transient
and reversible. However, such effects of As on hormone
signaling at key developmental or differentiation points might
be expected to result in long-term effects related to
epigenetic phenomena such as imprinting. In this regard, it is
interesting to note several recent studies that support this
idea (Liu et al. 2006a; Shen et al. 2006; Waalkes et al.
2004a). Among other interesting changes, a profound
up-regulation of ER expression has been reported in livers of
adult mice exposed in utero to As, in addition to their increased
incidence of liver tumors (Chen et al. 2004; Liu et al. 2006b;
Waalkes et
al. 2004b). Two recent epidemiology studies of humans exposed
to As in drinking water were also provocative (Smith et al.
2006; Wasserman et al. 2004). These studies suggest that there
can be significant long-term consequences of exposing young or
developing individuals to As during critical periods of
development. We propose that disruption of hormone signaling
is likely to be an important component of these effects. Thus,
understanding the role of As as an endocrine disruptor will be
important for assessing the overall impact of As on human
health and in developing appropriate risk assessment paradigms
that are protective of human health. |
|
|
| [References Listed in PubMed]
References
Abernathy CO, Liu YP, Longfellow
D, Aposhian HV, Beck BD, Fowler B, et al. 1999. Arsenic: health
effects, mechanisms of actions, and research issues. Environ
Health Perspect 107:593–597.
Abernathy CO, Thomas DJ,
Calderon RL. 2003. Health effects and risk assessment of arsenic.
J Nutr
133(suppl 1):1536S–1538S.
Abu-Abed S, Dolle P, Metzger
D, Beckett B, Chambon P, Petkovich M. 2001. The retinoic acid-metabolizing
enzyme, Cyp26A1, is essential for normal hindbrain patterning,
vertebral identity, and development of posterior structures.
Genes Dev 15:226–240.
Allen BM. 1916. The results of extirpation
of the anterior lobe of the hypophysis and the thyroid of Rana pipiens larvae.
Science 44:755–758.
Allen T, Rana SV. 2003.
Oxidative stress by inorganic arsenic: modulation by thyroid
hormones in rat.
Comp Biochem Physiol C Toxicol Pharmacol 135:157–162.
Andrew AS, Burgess JL, Meza MM, Demidenko
E, Waugh MG, Hamilton JW, et al. 2006. Arsenic exposure is
associated with decreased DNA repair in vitro and in
individuals exposed to drinking water arsenic. Environ Health
Perspect 114:1193–1198.
Andrew AS, Warren AJ, Barchowsky
A, Temple KA, Klei L, Soucy NV, et al. 2003. Genomic and proteomic
profiling of responses to toxic metals in human lung cells.
Environ Health Perspect 111:825–835.
Aposhian HV, Aposhian MM.
2006. Arsenic toxicology: five questions. Chem Res Toxicol 19:1–15.
Bastien J, Rochette-Egly
C. 2004. Nuclear retinoid receptors and the transcription of
retinoid-target
genes. Gene 328:1–16.
Bodwell JE, Gosse JA, Nomikos
AP, Hamilton JW. 2006. Arsenic disruption of steroid receptor
gene
activation: complex dose-response effects are shared by several
steroid receptors. Chem Res Toxicol 19:1619–1629.
Bodwell JE, Kingsley LA,
Hamilton JW. 2004. Arsenic at very low concentrations alters
glucocorticoid
receptor (GR)-mediated gene activation but not GR-mediated gene
repression: complex dose-response effects are closely
correlated with levels of activated GR and require a functional
GR DNA binding domain. Chem Res Toxicol 17:1064–1076.
Brown DD, Wang Z, Furlow JD, Kanamori A,
Schwartzmann RA, Remo BF, et al. 1996. The thyroid
hormone-induced tail resorption program during Xenopus laevis metamorphosis.
Proc Natl Acad Sci USA 93:1924–1929.
Buchholz DR, Hsia SC, Fu
L, Shi YB. 2003. A dominant-negative thyroid hormone receptor
blocks amphibian
metamorphosis by retaining corepressors at target genes. Mol
Cell Biol 23:6750–6758.
Buchholz DR, Paul BD, Fu L, Shi YB. 2006.
Molecular and developmental analyses of thyroid hormone
receptor function in Xenopus laevis, the African clawed
frog. Gen Comp Endocrinol 145:1–19.
Buckbinder L, Brown DD. 1993. Expression
of the Xenopus laevis prolactin and thyrotropin genes during
metamorphosis. Proc Natl Acad Sci USA 90:3820–3824.
Chen H, Li S, Liu J, Diwan
BA, Barrett JC, Waalkes MP. 2004. Chronic inorganic arsenic exposure
induces
hepatic global and individual gene hypomethylation:
implications for arsenic hepatocarcinogenesis. Carcinogenesis
25:1779–1786.
Das B, Cai L, Carter MG, Piao YL, Sharov
AA, Ko MS, et al. 2006. Gene expression changes at
metamorphosis induced by thyroid hormone in Xenopus laevis tadpoles.
Dev Biol 291:342–355.
Davey JC, Bodwell JE, Gosse JA, Hamilton
JW. 2007. Arsenic as an endocrine disruptor: effects of arsenic
on estrogen receptor-mediated gene expression in vivo and in
cell culture. Toxicol Sci 98:75-86.
Dodd MHI, Dodd JM. 1976. Physiology of the
Amphibia. New York: Academic Press.
Du Pasquier D, Rincheval V, Sinzelle L,
Chesneau A, Ballagny C, Sachs LM, et al. 2006. Developmental
cell death during Xenopus metamorphosis involves BID
cleavage and caspase 2 and 8 activation. Dev Dyn 235:2083–2094.
Galton VA. 2005. The roles
of the iodothyronine deiodinases in mammalian development. Thyroid
15:
823–834.
Gothe S, Wang Z, Ng L,
Kindblom JM, Barros AC, Ohlsson C, et al. 1999. Mice devoid of
all known thyroid
hormone receptors are viable but exhibit disorders of the
pituitary-thyroid axis, growth, and bone maturation. Genes Dev
13:1329–1341.
Gudernatch JF. 1912. Feeding
experiments on tadpoles. I. The influence of specific organs
given as food
on growth and differentiation: a contribution to the knowledge
of organs with internal secretion. Arch Entwicklungsmech Org
35:
457–483.
Hermanson O, Glass CK,
Rosenfeld MG. 2002. Nuclear receptor coregulators: multiple modes
of modification.
Trends Endocrinol Metab 13:55–60.
Hwang BJ, Utti C, Steinberg
M. 2006. Induction of cyclin D1 by submicromolar concentrations
of
arsenite in human epidermal keratinocytes. Toxicol Appl
Pharmacol 217:161–167.
Jakobs TC, Schmutzler C,
Meissner J, Kohrle J. 1997. The promoter of the human type I
5’-deiodinase gene—mapping of the transcription
start site and identification of a DR+4
thyroid-hormone-responsive element. Eur J Biochem 247:
288–297.
Kaltreider RC, Davis AM,
Lariviere JP, Hamilton JW. 2001. Arsenic alters the function
of the
glucocorticoid receptor as a transcription factor. Environ
Health Perspect 109:245–251.
Karagas MR, Tosteson TD,
Morris JS, Demidenko E, Mott LA, Heaney J, et al. 2004. Incidence
of
transitional cell carcinoma of the bladder and arsenic exposure
in New Hampshire. Cancer Causes Cont 15:465–472.
Kitagawa S, Obata T, Willingham
MC, Cheng SY. 1987. Thyroid hormone action: induction of morphological
changes and stimulation of cell growth in rat pituitary tumor
GH3 cells. Endocrinology 120:2591–2596.
Kitamura S, Jinno N, Ohta
S, Kuroki H, Fujimoto N. 2002. Thyroid hormonal activity of the
flame
retardants tetrabromobisphenol A and tetrachlorobisphenol A.
Biochem Biophys Res Commun 293:554–559.
Kitchin KT. 2001. Recent
advances in arsenic carcinogenesis: modes of action, animal model
systems,
and methylated arsenic metabolites. Toxicol Appl Pharmacol 172:
249–261.
Kurokawa R, DiRenzo J,
Boehm M, Sugarman J, Gloss B, Rosenfeld MG, et al. 1994. Regulation
of retinoid
signalling by receptor polarity and allosteric control of
ligand binding. Nature 371:528–531.
Lim W, Nguyen NH, Yang HY, Scanlan TS,
Furlow JD. 2002. A thyroid hormone antagonist that inhibits
thyroid hormone action in vivo. J Biol Chem 277:35664–35670.
Liu J, Xie Y, Ducharme DM, Shen J, Diwan
BA, Merrick BA, et al. 2006b. Global gene expression associated
with hepatocarcinogenesis in adult male mice induced by in utero arsenic
exposure. Environ Health Perspect 114:404–411.
Liu J, Xie Y, Merrick BA,
Shen J, Ducharme DM, Collins J, et al. 2006a. Transplacental
arsenic plus
postnatal 12-O-teradecanoyl phorbol-13-acetate exposures
associated with hepatocarcinogenesis induce similar aberrant
gene expression patterns in male and female mouse liver.
Toxicol Appl Pharmacol 213:216–223.
Loudig O, Babichuk C, White J, Abu-Abed S,
Mueller C, Petkovich M. 2000. Cytochrome P450RAI (CYP26)
promoter: a distinct composite retinoic acid response element
underlies the
complex regulation of retinoic acid metabolism. Mol Endocrinol
14:1483–1497.
Mann KK, Padovani AM, Guo
Q, Colosimo AL, Lee HY, Kurie JM, et al. 2005. Arsenic trioxide
inhibits
nuclear receptor function via SEK1/JNK-mediated RXRalpha
phosphorylation. J Clin Invest 115:2924–2933.
Mark M, Ghyselinck NB,
Chambon P. 2006. Function of retinoid nuclear receptors: lessons
from genetic
and pharmacological dissections of the retinoic acid signaling
pathway during mouse embryogenesis. Annu Rev Pharmacol Toxicol
46:451–480.
McKenna NJ, O’Malley BW. 2002.
Minireview: nuclear receptor coactivators—an update.
Endocrinology 143:2461–2465.
Miyazaki K, Watanabe C,
Mori K, Yoshida K, Ohtsuka R. 2005. The effects of gestational
arsenic exposure
and dietary selenium deficiency on selenium and selenoenzymes
in maternal and fetal tissues in mice. Toxicology 208:
357–365.
Mukherjee A, Sengupta MK,
Hossain MA, Ahamed S, Das B, Nayak B, et al. 2006. Arsenic contamination
in groundwater: a global perspective with emphasis on the Asian
scenario. J Health Popul Nutr 24:142–163.
Nakajima K, Yaoita Y. 2003.
Dual mechanisms governing muscle cell death in tadpole tail during
amphibian metamorphosis. Dev Dyn 227:246–255.
National Library of Medicine. 2007.
GenBank Overview. Available: http://www.ncbi.nlm.nih.gov/Genbank/ [accessed 27 November 2007].
Nieuwkoop PD, Faber J. 1994. Normal Table
of Xenopus laevis (Daudin). A Systematical and Chronological
Survey of the Development from the Fertilized Egg till the End
of Metamorphosis. London:Garland Publishing, Inc.
NRC (National Research Council). 1999.
Arsenic in Drinking Water. Washington, DC:National Academy
Press.
NRC (National Research Council). 2001.
Arsenic in Drinking Water: 2001 Update. Washington DC:National
Academy Press.
Oppenheimer JH, Samuels HH. 1983.
Molecular Basis of Thyroid Hormone Action. New York:Academic
Press.
Porterfield SP, Hendrich
CE. 1993. The role of thyroid hormones in prenatal and neonatal
neurological
development—current perspectives. Endocr Rev 14:
94–106.
Raz Y, Kelley MW. 1997.
Effects of retinoid and thyroid receptors during development
of the inner
ear. Sem Cell Dev Biol 8:257–264.
Rossman TG. 2003. Mechanism
of arsenic carcinogenesis: an integrated approach. Mutat Res
533:
37–65.
Shaw JR, Gabor K, Hand E, Lankowsky A,
Durant L, Thibideau R, et al. 2007. Role of glucocorticoid
receptor in acclimation of killifish (Fundulus heteroclitus)
to seawater and effects of arsenic. Am J Physiol Regul Integr
Comp
Physiol 292:R1052–1060.
Shen J, Liu J, Xie Y, Diwan
BA, Waalkes MP. 2006. Fetal onset of aberrant gene expression
relevant to
pulmonary carcinogenesis in lung adenocarcinoma development
induced by in utero arsenic exposure. Toxicol Sci 89:
108–119.
Shenefelt RE. 1972. Morphogenesis
of malformations in hamsters caused by retinoic acid: relation
to
dose and stage at treatment. Teratology 5:103–118.
Shi YB, Ritchie JW, Taylor
PM. 2002. Complex regulation of thyroid hormone action: multiple
opportunities for pharmacological intervention. Pharmacol Ther
94: 235–251.
Smith AH, Hopenhayn-Rich
C, Bates MN, Goeden HM, Hertz-Picciotto I, Duggan HM, et al.
2002. Cancer
risks from arsenic in drinking water. Environ Health Perspect
97:259–267.
Smith AH, Marshall G, Yuan Y, Ferreccio C,
Liaw J, von Ehrenstein O, et al. 2006. Increased mortality from
lung cancer and bronchiectasis in young adults after exposure
to arsenic in utero and in early childhood. Environ
Health Perspect 114:1293–1296.
Stanton CR, Thibideau R, Lankowsky A, Shaw
JR, Hamilton JW, Stanton BA. 2006. Arsenic inhibits
CFTR-mediated chloride secretion by killifish (Fundulus heteroclitus)
opercular membrane. Cell Physiol Biochem 17: 269–278.
Tapio S, Grosche B. 2006.
Arsenic in the aetiology of cancer. Mutat Res 612:215–246.
Tata JR. 1966. Requirement
for RNA protein synthesis for induced regression of the tadpole
tail in organ
culture. Dev Biol 13:77–94.
Thompson JN, Howell JM,
Pitt GA. 1964. Vitamin A and reproduction in rats. Proc R Soc
Lond B Biol Sci
159:510–535.
Tsai M-J, O’Malley BW. 1994.
Molecular mechanisms of action of steroid/thyroid receptor
superfamily members. Annu Rev Biochem 63:451–486.
Veldhoen N, Helbing CC. 2001. Detection of
environmental endocrine-disruptor effects on gene expression
in live Rana catesbeiana tadpoles using a tail fin biopsy
technique. Environ Toxicol Chem 20:2704–2708.
Vogt BL, Rossman TG. 2001.
Effects of arsenite on p53, p21 and cyclin D expression in normal
human
fibroblasts—a possible mechanism for arsenite’s
comutagenicity. Mutat Res 478:159–168.
Waalkes MP, Liu J, Chen
H, Xie Y, Achanzar WE, Zhou YS, et al. 2004b. Estrogen signaling
in livers of male
mice with hepatocellular carcinoma induced by exposure to
arsenic in utero. J Natl Cancer Inst 96:466–474.
Waalkes MP, Ward JM, Diwan
BA. 2004a. Induction of tumors of the liver, lung, ovary and
adrenal in
adult mice after brief maternal gestational exposure to
inorganic arsenic: promotional effects of postnatal phorbol
ester exposure on hepatic and pulmonary, but not dermal
cancers. Carcinogenesis 25:133–141.
Waalkes MP, Ward JM, Liu
J, Diwan BA. 2003. Transplacental carcinogenicity of inorganic
arsenic in
the drinking water: induction of hepatic, ovarian, pulmonary,
and adrenal tumors in mice. Toxicol Appl Pharmacol 186:
7–17.
Wasserman GA, Liu X, Parvez
F, Ahsan H, Factor-Litvak P, van Geen A, et al. 2004. Water arsenic
exposure and children’s intellectual function in
Araihazar, Bangladesh. Environ Health Perspect 112:
1329–1333.
Watanabe C, Inaoka T, Matsui
T, Ishigaki K, Murayama N, Ohtsuka R. 2003. Effects of arsenic
on younger
generations. J Environ Sci Health 38(Part A):129–139.
White JA, Beckett-Jones
B, Guo YD, Dilworth FJ, Bonasoro J, Jones G, et al. 1997. cDNA
cloning of
human retinoic acid-metabolizing enzyme (hP450RAI) identifies
a
novel family of cytochromes P450. J Biol Chem 272:
18538–18541.
White JA, Guo YD, Baetz
K, Beckett-Jones B, Bonasoro J, Hsu KE, et al. 1996. Identification
of the
retinoic acid-inducible all-trans-retinoic acid 4-hydroxylase.
J Biol Chem 271:29922–29927.
Wolbach SB, Howe PR. 1978.
Nutrition classics. The Journal of Experimental Medicine 42:
753–777, 1925. Tissue changes following deprivation of
fat-soluble A vitamin. S. Burt Wolbach and Percy R. Howe. Nutr
Rev 36:16–19.
Wong J, Shi YB. 1995. Coordinated
regulation of and transcriptional activation by Xenopus thyroid
hormone and retinoid X receptors. J Biol Chem 270:
18479–18483.
Yamamoto S, Konishi Y,
Matsuda T, Murai T, Shibata MA, Matsui-Yuasa I, et al. 1995.
Cancer induction
by an
organic arsenic compound, dimethylarsinic acid (cacodylic
acid), in F344/DuCrj rats after pretreatment with five
carcinogens. Cancer Res 55:1271–1276.
Yaoita Y, Brown DD. 1990.
A correlation of thyroid hormone receptor gene expression with
amphibian
metamorphosis. Genes Dev 4(11):1917–1924.
Zhang J, Lazar MA. 2000.
The mechanism of action of thyroid hormones. Annu Rev Physiol
62:439–466. |
|
|
|
| |













