Address correspondence to J. D. Roberts, MD C2-14, NIEHS, PO Box 12233, Research Triangle Park, NC 27709 USA.
Over 250 people participated in a 2-day conference, "Molecular Mechanisms of Environmental Carcinogenesis," at the National Institute of Environmental Health Sciences (NIEHS) in Research Triangle Park, North Carolina, 19-20 September 1994. The purpose of the conference was to bring together scientists to discuss recent advances in basic cancer research, with an emphasis on the contribution of environmental factors to neoplasia. The conference was initiated with a brief welcoming address by NIEHS Director Kenneth Olden, and included 17 platform talks as well as a collection of poster presentations.
The conference was divided into four sessions. The first session focused on cell cycle control and cancer. Normal cells carefully monitor cell cycle events such as DNA replication and cell division to maintain a stable genome. Failure to regulate the cell cycle appropriately is a fundamental characteristic of cancer cells. Elucidation of the molecular events involved in cell cycle transitions, genetic instability, and responses to DNA damage is critical for understanding the impact of the environment on carcinogenesis.
The fission yeast, Schizosaccharomyces pombe, has provided an important model system for the identification of checkpoint genes. Mutants of S. pombe that fail to undergo cell cycle arrest when DNA synthesis is inhibited by hydroxyurea have recently been identified. These mutants proceed through mitosis and cell division without completing DNA replication. Because the molecules that control cell cycle transitions in yeast have been conserved through evolution, it will be of interest to elucidate the function of these new genes to ascertain their roles, if any, in mammalian cell checkpoint control.
The molecular details of how oncogenes and tumor-suppressor genes function in growth control are becoming clearer from studies of the retinoblastoma susceptibility gene product (pRB) and the product of the c-abl tyrosine kinase proto-oncogene, p170Abl. Dominant-negative mutations in Rb disrupt the interactions between pRb and p170Abl, resulting in an inhibition of the growth suppression function of pRb. Recently, mutations have been generated in Rb that prevent this growth-suppression function from being blocked by phosphorylation. These and other results have been used to suggest that p170Abl can regulate pRb complexes that in turn regulate entry into and exit from the cell cycle, as well as progression through DNA synthesis.
The role of the p53 tumor-suppressor gene product in cancer is of particular interest because p53 is the most commonly mutated gene in human cancers characterized to date. Cells that lack a functional p53 cannot undergo cell cycle arrest in response to DNA damage and will enter DNA synthesis with unrepaired DNA. The molecular dynamics of the cellular response pathways that involve p53 in response to DNA damage are currently under intense scrutiny. DNA strand breaks, induced by either ionizing radiation or microinjection of a restriction endonuclease, have been shown to lead to an increase in p53. This increase requires the currently undefined gene product(s) that are defective in ataxia-telangiectasia, a cancer-prone inherited disease. The induction of p53 results in either cell cycle delay or initiation of programmed cell death (apoptosis), depending on the cell type. A practical application of a better understanding of this pathway might be the specific induction of apoptosis in tumor cells after exposure to therapeutic agents.
Cytoskeletal alterations and genomic instability in cells transformed by the mos oncogene were also discussed in the first session. Murine fibroblasts infected with a mos virus have elevated levels of mitogen-activated protein (MAP) kinase activity and displaced spindles that are abnormally attached to the cell membrane during mitosis. The alteration in cytoskeletal components in mos-transformed cells, as well as inappropriate expression of certain mitotic phenotypes in interphase cells, could contribute to genomic instability in these cells, thus providing a model system for the further study of genomic instability in tumors.
The second session focused on cancer susceptibility genes. For many years, it has been clear that certain individuals and their offspring are prone to develop specific types of cancers at an earlier age or with an increased frequency compared to the general population. With recent improvements in DNA mapping strategies, along with advances in molecular biological techniques, scientists now can identify defects in specific genes that are responsible for these familial cancers. Among the relevant topics addressed at this conference were epidemiological studies of cancer prone families, characterization of a recently identified breast cancer gene, the role of heritable differences in xenobiotic metabolism in cancer susceptibility, and mouse models for the identification of new cancer susceptibility genes.
Data from kindreds of patients who develop soft tissue sarcomas early in life revealed strong evidence for a dominantly inherited cancer gene. Some of the patients were characterized as having Li-Fraumeni syndrome; their disease could be linked to mutations in the p53 tumor-suppressor gene. Recent studies have suggested an earlier age of cancer onset in consecutive generations of families carrying p53 mutations. This interesting observation could be due to improved screening and detection methods, an increase in environmental insults, or genetic instability associated with mutations in p53. Better monitoring and early detection of tumors will be critical for families with mutations predisposing them to cancer and should provide valuable insight into environmental carcinogenesis. Two presentations focused on the highly publicized breast cancer gene, BRCA1. One provided a historical context of the identification of the BRCA1 gene and its role in the development of breast and ovarian cancers. BRCA1 is thought to account for at least 85% of families with increased incidence of both early onset breast cancer and ovarian cancer. However, less than half the families with site-specific breast cancer are linked to BRCA1. Several of the site-specific breast cancer families that are not associated with BRCA1 are linked to a second breast cancer susceptibility gene, BRCA2, which has recently been localized to chromosome 13q. A second presentation described work performed by scientists at NIEHS in collaboration with scientists at the University of Utah and Myriad Genetics on the molecular cloning of BRCA1 and the characterization of mutations in the gene. The BRCA1 gene encodes a relatively large protein of 1863 amino acids with a RING finger motif in its amino-terminal region. Characterization of BRCA1 may provide valuable insights into breast cancer biology.
The interaction of pro-carcinogens and genetic variability was stressed in several presentations. These included a discussion of allelic differences in the activation of chemical carcinogens by human cytochromes P450 (CYP450) and glutathione S-transferases with regard to their role in cancer susceptibility. Individuals with certain CYP450 alleles may be more susceptible to metabolic activation of carcinogens. The combination of genetic variation and environmental exposure represents one example of the complexity evident in carcinogenesis, from initial exposures to environmental pro-carcinogens to the establishment of genetically unstable tumors with highly malignant phenotypes. Identification of polymorphisms in xenobiotic metabolism alleles associated with increased incidence of cancer may enable researchers to evaluate individual exposure risks. One approach to unraveling these complex interactions is to use animal models in which the combination of genetic background and environmental exposure can be controlled. Mouse models have also been used to dissect tumor-susceptibility genes involved in hepatocarcinogenesis. For example, mice treated with the mutagen N,N-diethylnitrosamine showed a 100-fold difference in tumor multiplicity depending on which inbred strain was used. Such a strategy provides an experimental approach for identifying multiple genetic loci controlling the susceptibility to liver tumors. At the meeting, it was proposed that the promotion phase of hepatocarcinogenesis can be divided into a conversion and a proliferation step. The use of genetic studies to identify genes crucial for the development of tumors should improve our understanding of environmental carcinogenesis.
The third session introduced the importance of hormones in carcinogenesis, with an emphasis on estrogens. It is now clear that estrogens cause cell proliferation in certain in vitro systems and enhance tumor formation in carcinogen-exposed animals. However, estrogen effects in humans vary by tissue type, exposure pattern, and subject group. The relative risks of endometrial and breast cancer after estrogen exposure was a topic of great interest. Many women are now exposed to estrogens, through oral contraceptives and estrogen replacement therapy, in a variety of doses and admixtures, throughout their lives. This long-term exposure makes the assessment of relative risk for any single exposure much more difficult. Furthermore, it was pointed out that the baseline cancer rate in women changes with age, and it is critical to take such changes into account when judging the significance of any apparent increase in relative risk.
The pathway by which estrogen increases cancer risk was also addressed. Important advances have been made in defining the genetic alterations that characterize estrogen-associated cancers, focusing on endometrial carcinoma and clear-cell adenocarcinoma of the vagina and cervix resulting from exposure to diethylstilbestrol in utero. In addition to DNA sequencing of tumor-suppressor genes, a search for loss of heterozygosity (LOH) has been used to identify critical genetic elements. Clinical data have indicated that LOH for human chromosome 14q is closely associated with a poor prognosis in patients with endometrial carcinoma. One hope is that positional cloning will help narrow the search for the genes involved. Another set of genes now thought to be critical to the regulation of genome stability are those involved in DNA mismatch repair. Microsatellite instability, observed in a variety of tumor cells, also appears to be present in as much as 10-20% of human breast carcinomas. An analysis of human mismatch repair genes, such as MSH2, is underway to see if inactivation of key repair proteins is responsible for genetic instability in various neoplasms.
Recent research has also explored estrogen-mediated changes in the control of proliferation by developing a series of variants of the human breast cell line, MCF7. A factor in serum, called estrocolyone-1, has been identified that specifically inhibits the proliferation of estrogen-sensitive cells. Variants of MCF7 have been generated that are no longer inhibited by serum, but are inhibited by estradiol. One of the implications of this work for therapy is that the selective manipulation of a patient's endocrine milieu can be important for regulating growth of estrogen-sensitive tumors.
The recognition of prostate cancer as a health issue for men has increased greatly in the last decade: current estimates are that 90% of men at age 80 will have some level of hyperplastic prostate disease. Research is in progress on the genetics and cell biology of prostatic tumors, with the goal of developing a set of tumor markers that can be used reliably to diagnose the severity of the disease. A model was proposed in which a set of proteins involved in cell-cell contact is inactivated, leading to a loss of adhesion and increased metastasis. These proteins include E-cadherin, 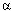 -catenin, and ß-catenin. Consistent with this model, the introduction of chromosome 5 into a prostatic tumor cell line, with a consequent expression of -catenin, led to an increase in cell-cell contact. An analysis of families manifesting higher incidences of prostatic cancer, combined with an analysis of metastasis-suppressor genes in animal models, should enhance the ability to identify critical genes and to predict the potential severity of disease discovered in the early stages.
-catenin, and ß-catenin. Consistent with this model, the introduction of chromosome 5 into a prostatic tumor cell line, with a consequent expression of -catenin, led to an increase in cell-cell contact. An analysis of families manifesting higher incidences of prostatic cancer, combined with an analysis of metastasis-suppressor genes in animal models, should enhance the ability to identify critical genes and to predict the potential severity of disease discovered in the early stages.
The final session of the meeting focused on tumor-suppressor genes and their biochemical functions. Studies of pRB and its interactions with the E1A protein of adenovirus have provided clues to the functions of these proteins in viral transformation. The E1A protein has been found to bind pRB and displace the E2F transcription factor that is usually bound by pRB. E2F DNA binding sites have been identified in several genes known to be involved in growth control. Thus, we can now postulate mechanisms for how environmentally induced mutations or viral infection can lead to a loss of growth control through the disruption of specific protein-protein interactions. Furthermore, the cloning of E2F homologues from Drosophila melanogaster has allowed an extensive analysis of E2F protein structure and expression during embryogenesis and may provide a useful marker for cells that have recently undergone cell division.
It is clear that animal models will continue to be important tools for studying the initiation and progression of cancers. For example, a simple base change that leads to a murine mutation, min (multiple intestinal neoplasia), predisposes mice to intestinal and mammary tumors. However, the genetic background of the mouse in which min is studied significantly alters the expression of neoplasia. Genetic loci termed mom, for modifier of min, have been identified and mapped to mouse distal chromosome 4. The influence of carcinogens on the incidence of neoplasia in strains with different genetic backgrounds is underway and should allow a more complete understanding of the interplay between genetic predisposition and environmental exposure.
Mutational spectra of tumor-suppressor genes are being studied to reveal the underlying patterns of spontaneous mutation in these genes and the biological relevance of specific mutations. Studies of a large number of mutations in the Rb and p53 genes indicated that the vast majority of Rb mutations were nonsense errors, whereas p53 mutations were diverse and varied according to the tissue of origin. One class of p53 G-to-T transversion mutations was overrepresented and was postulated to be the result of environmental mutagen exposure. However, phenotypic selection for relevant mutations may vary in different organs, and methodological biases must be accounted for before these spectra can be used to shed light on the role of specific mutations in the etiology of cancer.
The importance of repair processes was reiterated in a discussion of human mutators hMSH2 and hMLH1. The presence of multiple mutated genes in tumor cells has led to the question of how cancer cells arise given the usual low mutation rates in eukaryotic cells. A number of proposals have been made as to which kinds of replication and repair functions might be inactivated to generate mutator phenotypes. hMSH2 and hMLH1 belong to highly conserved families of mismatch repair proteins. Defects in these genes lead to increases in mutation frequencies of up to 1000-fold and enhanced rates of recombination. In some tumors, including the hereditary nonpolyposis colon cancer syndrome, mutator phenotypes due to aberrant repair may explain the observed increases in microsatellite instability. Alterations in other mutator genes may also play a critical role in tumor development.
In summary, this conference provided a strong reminder of the complexity of cancer, a disease with both environmental and genetic risk factors. The excitement generated by the discovery of new genes linked to familial disease was tempered by the realization that any single gene may be directly involved in a small percentage of cancers. However, it may be expected that the molecular and biochemical analysis of such genes will provide insights into the pathways by which processes such as cell growth and cell-cell contact are disrupted. Such an understanding should allow researchers to seek out, in a logical fashion, other candidate genes as well as environmental agents that influence these pathways. Finally, a more complete description of how these pathways are disrupted in cancer cells may provide the knowledge to develop effective preventive and therapeutic strategies, based on clear biochemical mechanisms and, ultimately, an improvement in our ability to successfully treat patients with cancer.
Molecular Mechanisms of Environmental Carcinogenesis
Sponsors:
National Institute of Environmental Health Sciences
Burroughs Wellcome Co.
Chemical Industry Institute of Toxicology
Genotoxicity and Environmental Mutagen Society
Glaxo Research Laboratories, Glaxo, Inc.
North Carolina Biotechnology Center
The Proctor and Gamble Company
Organizers:
Richard S. Paules (chair), J. Carl Barrett, Kenneth W. Batchelor,
Jeffrey A. Boyd, R. Julian Preston, Kenneth R. Tindall, Roger W. Wiseman
Speakers:
Jeffrey A. Boyd, Norman R. Drinkwater, Nick Dyson, Tamar Enoch,
Richard Fishel, Andrew Futreal, Frank J. Gonzalez, Barbara S. Hulka,
William B. Isaacs, Michael B. Kastan, Amy R. Moser, Steven Narod,
Ana M. Soto, Louise C. Strong, George F. Vande Woude, Jean Y.
J. Wang, David W. Yandell
|
[Table of Contents]
Last Update: August 5, 1997





