FDA > CDRH >
CDRH Learn > Presentation: 510(k)�Overview
Presentation: 510(k) Overview
(This presentation is also available in video format with captioning and PDF printer-friendly format.)
Heather Rosecrans
Director, 510(k) Staff
Center for Devices & Radiological Health, FDA
Medical Device Amendments of 1976 �to the FF,D,&C Act
- May 28, 1976
- Defined a device (201(h) of the Act)
- Required classification of device types legally on the market at that time
- Led to classification of approximately 1,700 different generic types of devices and grouped them into 19 medical specialties
- Required premarket review of devices
What is a 510(k)
- Premarket Notification
- Section 510(k) of Federal Food, Drug, & Cosmetic Act
- 21 CFR 807 Subpart E
- Marketing Clearance Application
- Allows FDA to Determine Substantial Equivalence (SE)
- “The” classification process for a device
What a 510(k) Is Not
- A Form
- Establishment Registration
- Device Listing
- Premarket Approval (PMA)
510(k) & Classification
- A 510(k) is the classification process for individual�post-amendment devices by:
- Finding the device substantially equivalent (SE) or�
- Finding the device not substantially equivalent (NSE)
Pre-amendment vs. Post-amendment
The F F, D, & C Act divided the arena of medical devices depending on when the devices were introduced into commercial distribution:
- Pre-amendments Devices (pre-May 28, 1976)
- Exempted (with conditions) from marketing clearance
VS.
- Post-amendments Devices (post-May 28, 1976)
- Requires marketing clearance
What is a Predicate?�21CFR Part 807.92(a)(3)
An identification of the legally marketed device to which the submitter claims equivalence. A legally marketed device to which a new device may be compared for a determination regarding substantial equivalence is��
a device that was legally marketed prior to May 28, 1976, or a device which has been reclassified from class III to class II or I (the predicate),� �
or a device which has been found to be substantially equivalent through the 510(k) premarket notification process;
What is a Device Type? �
- 21 CFR 860.3(i)
- Generic type of device means a grouping of devices that do not differ significantly in purpose, design, materials, energy source, function, or any other feature related to safety and effectiveness, and for which similar regulatory controls are sufficient to provide reasonable assurance of safety and effectiveness.
Regulatory Classes
Three Regulatory Classes (level of control based on risk):
- Class I – General Controls
- Class II – General Controls & Special Controls
- Class III – General Controls and Premarket Approval
Classification
- Classification regulations for individual device types are found in 21 CFR Parts 862-892
- Regulations describe the device type as it existed Prior to May 28, 1976
- New indications for use or new technologies are assigned new product codes
- Classification regulations for individual device types found in 21 CFR Parts 862-892
Example:
Part 872 – Dental Devices
138 Device Types, e.g.,:
Sec. 872.4760 Bone plate. (a) Identification. A bone plate is a metal device intended to stabilize fractured bone structures in the oral cavity. The bone segments are attached to the plate with screws to prevent movement of the segments. (b) Classification. Class II.
Substantially Equivalent (SE) Ltr
Classification Regulation appears on all SE Letters and are available on the Internet
510(k) Exempt Devices
- Preamendments Devices
- Unfinished Devices
- Devices Exempt by Statute or by regulation from 510(k)
739 Class I (93%), 74 Class II (8%)
- Finished Devices not Sold in U.S.
- Devices Covered Under Another 510(k), e.g., Private Labeled Device
- Custom Devices
- Veterinary Devices
Limitations of Exemption �from 510(k) - Class I & II
- Found in “.9” of Classification Chapters
- Four Limitations
- If the device has an intended use that is different from the intended use of a legally marketed device in that generic type
- Operates using a different fundamental scientific technology than that used by a legally marketed device in that generic type
- Specific limitations for in vitro diagnostic devices
- Device specific limitations as specified in a classification regulation
Regulatory Class
- Class determines type of premarket submission required by FDA
- Class I or II 510(k) Exempt
- Subject to limitations on exemptions covered under 21 CFR xxx.9 (e.g., 862.9 to 892.9)
- Class I or II Non 510(k) Exempt
- Class III
- PMA (510(k) for pre-amendment devices until 515(b) calls for PMA or the device type is reclassified)
510(k) Submission �Required When?
- Introducing a device to the market for the first time
- Change in indications for use for a previously cleared device
- Making significant modification to a previously cleared device
Who Must Submit a 510(k)?
- Manufacturers
- Specifications Developers
- Repackagers who change device or its labeling
- Relabelers who change the labeling- e.g., instructions for use
- Anyone who both manufactures & distributes
Who is Not Required �to Submit a 510(k)?
Private Label Distributor who ONLY adds company name & wording such as: �
“Distributed by ___________” or�
“Manufactured for _________”
- Repackager who does not alter labeling
- Distributor or Importer who furthers �marketing of the legally marketed device and does not alter labeling or change device
Information Requested in 510(k) �(21 CFR § 807.87)
- Submitter’s name, address, phone & fax, contact person, rep/consultant name
- Device Regulation (Classification) Name, CFR number, device class, product code
- Common/usual name & trade/proprietary name & model number
- Indications for Use Statement
- Truthful and Accurate Statement
- Proposed labeling
- Adherence to voluntary standard and standard form
- Financial Certification or Disclosure Statement or both
- Identification of marketed device(s) to which equivalence is claimed
- Compliance with section 514 Special Controls
- Proposed labels, labeling, including any promotional material
- Photographs, engineering drawings
- Substantially equivalent statement & comparison with predicate
- Statement of similarities and/or differences with predicate device
- Data for changes for modified devices
- 510(k) MUST include either:
- 510(k) Statement (21 CFR 807.93)
510(k) Holder provides copy of 510(k) deleting trade secret & commercial confidential information to anyone within 30 days, OR
- 510(k) Summary (21 CFR 807.92)
FDA provides 510(k) summary, as provided by 510(k) Holder, to any requester and is available on our website
- Class III 510(k) must include: *Content and Format (21 CFR § 807.94)
- Certification & literature search has been conducted, and
- Summary of adverse S & E data with citation to the literature
- Performance Data (bench, animal, and/or clinical)�
- Sterilization, Software & Hardware Information, if any�
- Address information requested in specific guidance documents
Clinical Data in 510(k)
- Approximately 10% of all 510(k)s
- Important difference with the predicate device, e.g., new indication for use or new technology
- Must be collected under Investigational Device Exemption Regulations (21 CFR Part 812)
A 510(k) Must Contain:
- Proposed labeling sufficient to describe the device’s indications for use
- A description of how the device is similar to or different from other devices of comparable type (predicate device)
- Any other information the Center needs to determine whether the device is SE
FDA Requests �Additional Information-
- Administratively incomplete submissions
- When information/performance data are required to demonstrate equivalence
- Reviewer requests by telephone, email or letter
- Clock stops by letter only
- Submit to Document Mail Center
- 30 days to submit
- May request extension of time
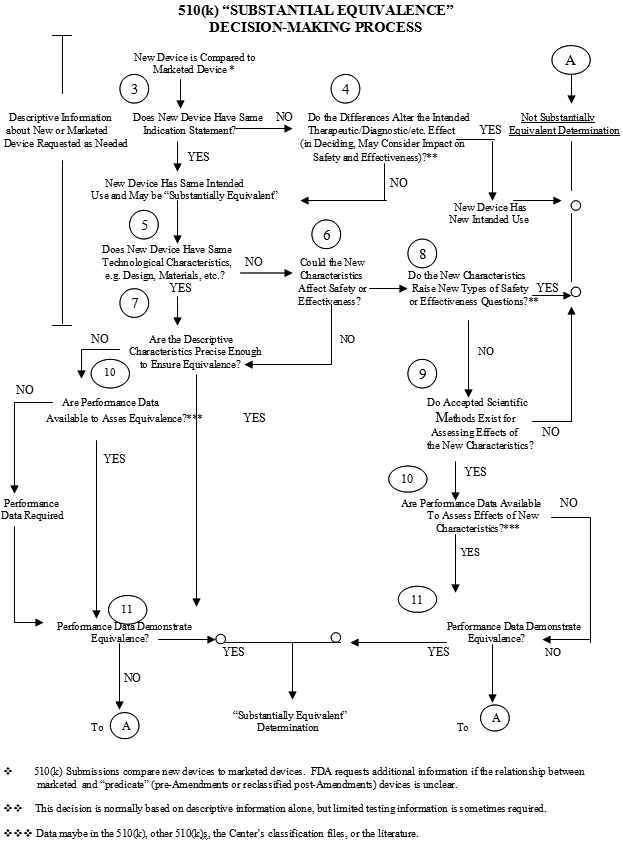
Establish Equivalence to:
- A legally marketed device (a predicate*) that does not require a PMA, i.e., a:
- Preamendments device*
- A device found by FDA to be Substantially Equivalent (SE), or
- A reclassified device*
*21 CFR 807.92(a)(3)
Device is �Substantially Equivalent (SE)
- If:
- In Comparison to a legally marketed device, it:
- Has the same intended use, and
- Has the same technological characteristics as the predicate device,
- Or . . . . . . . . . . . . .
- Has the same intended use, and
- Has different technological characteristics and the information in the 510(k):
- Does not raise new questions of safety and effectiveness, and
- Demonstrates it is at least as safe and effective as the predicate
FDA finds the device Substantially Equivalent (SE)?
- NO - PMA, Application or De Novo�
- YES - To Market
Not Substantially Equivalent (NSE)
- There is no predicate device
- Has a NEW intended use
- Has different technological characteristics compared to the predicate device and �raises a new type question of safety and effectiveness
- All examples above required no review �of data and will require PMA or De Novo
- Does not demonstrate that it is at least as safe and effective as the predicate
- The example above required review of data and is eligible for a new 510(k) with new data
- Approximately 3% are found NSE
- Data is looked at last in the 510(k) regulatory process
- FDA usually asks for additional information at least once prior to determining the device is NSE for lack of data
510(k) & Classification
- Finding the device not substantially equivalent (NSE)
- automatically places device into class III �and requires:
- PMA; or
- Reclassification before marketing
Modifications
Licensing of 510(k)s
- A firm may not BOTH manufacture and distribute a device without their own 510(k)
- (21 CFR 807.85(b)(2))
Finished Device� 820.3(l) – Quality System Regulation �
- 820.3(l) Finished device means any device or accessory to any device that is suitable for use or capable of functioning, whether or not it is packaged, labeled, or sterilized.
- Finished device in final form for sale to an end user is subject to 510(k) requirements
"Unfinished Device"�for purposes of 510(k)
- If not in final form, OR in final form but NOT for sale to an end user, is not subject to the 510(k) requirements
Accessories & Components
- Component (820.3(c)) - Component means any raw �material, substance, piece, part, software, firmware, �labeling, or assembly which is intended to be included �as part of the finished, packaged, and labeled device. �
- Accessory—”extras” (not defined in the regulations)�
- Accessories/components to a device take on the same classification as the "parent" device unless they are �separately classified �
- A finished accessory or a finished component sold to�an end user, is subject to 510(k) requirements.
Confidentiality of Information
21 CFR § 807.95
Misbranding by �Reference to 510(k)
21 CFR § 807.97
510(k) Overview
Heather Rosecrans
240-276-4021
heather.rosecrans@fda.hhs.gov
Updated December 4, 2008

CDRH Home Page | CDRH A-Z Index | Contact CDRH | Accessibility | Disclaimer
FDA Home Page | Search FDA Site | FDA A-Z Index | Contact FDA | HHS Home Page
Center for Devices and Radiological Health / CDRH




