
17QUALITY SYSTEM AUDITS
INTRODUCTION
AUDIT REQUIREMENTS
Procedure
Audit Schedule
Independent Auditor
Employee Training
Evaluation Criteria
Results and Corrective Actions
Audit Certification
EXHIBITS
Policy/Procedure for Quality System Audit
Quality System Audit Procedure
Vendor Survey Form
Section 820.20 outlines the quality system
requirements of the Quality System (QS) regulation. As discussed
in Chapter 2, every quality system should include: management
policies; objectives; an organization; documentation; performance
of tasks according to policies; monitoring of the system (feedback)
and corrective action as indicated by the feedback. Section 820.22
requires that the quality system be monitored through audits.
The analysis and use of feedback data from product acceptance,
audits, complaints, repairs, and other sources are necessary
parts of a self correcting quality system. Thus, the audit of
a quality system is one of the most important GMP requirements.
The quality system first implemented by a new manufacturer will
change as the manufacturer grows and as the company=s
products, operations and employees change. Therefore, a quality
system should change with the company. Quality system audits are
the primary tool for assuring that the quality system changes
are correct and are correctly implemented.
A quality audit is a documented independent
inspection and review of a quality system. The audit is performed
on a periodic basis in accordance with written procedures. The
objective is to verify, by examination and evaluation of objective
evidence, the actual degree of compliance with those elements
of the quality system under review. These audits are an essential
part of every medical device manufacturer's effort to assure safe
and effective devices. Regardless of how well a quality system
is planned, monitoring of the system is required if the quality
system program is to be effective in assuring that finished devices
meet specifications. FDA analysis of factory inspections has shown
that manufacturers who do not have an adequate quality audit system
usually do not have an adequate quality system. An evaluation
of approximately 2400 manufacturers that had received GMP inspections
by FDA showed that manufacturers with an adequate quality audit
system were in compliance with approximately 96 percent of the
GMP requirements, while those that did not have an adequate audit
system were in compliance with approximately 70 percent of the
requirements.
If conducted properly, a quality audit can
detect quality system defects. Isolation of unsatisfactory trends
and correction of factors that cause defective products prevent
the production of unsafe or nonconforming devices. Without an
effective quality audit function, the quality system program is
incomplete -- there is no assurance that a manufacturer is consistently
in a state-of-control. In addition, the proper implementationof a quality audit system can result in cost savings by identifying
and correcting problem areas. Without an audit, the quality system
becomes an open loop without feedback to management and without
corrective action. Without overt management support, the quality
system program will eventually become ineffective and, as history
has shown, ignored.
The QS regulation requires that planned
and periodic audits of the quality system shall be performed to
verify compliance with the quality system requirements. The audits
are to be performed in accordance with written procedures by appropriately
trained individuals who do not have direct responsibility for
the matters being audited. Audit results shall be documented in
written audit reports, which shall be reviewed by management personnel,
who have responsibility for the matters audited, and by other
involved parties. Follow-up corrective action, including re-audit
of deficient matters, shall be taken when indicated. Upon request
of a designated FDA employee, an employee in management with executive
responsibility shall certify in writing that the audits have been
performed and documented, the dates on which they were performed,
and that any required corrective action has been undertaken.
To assure that company quality goals will
be routinely met and to comply with the QS regulation, quality
system audits should:
Where practical, manufacturers should include
audits of their suppliers, calibration laboratories, and contractors
as part of a quality system audit. Manufacturers should audit
suppliers where needed, to assure that they have adequate quality
system controls for raw materials and components shipped to and
received by the manufacturers under supplier certification or
certificate of compliance with specifications. An example of a
detailed supplier (vendor) survey (audit checklist) form is in
the Exhibits.
All manufacturers should have a written
quality audit procedure, although the details will vary with the
manufacturer size and nature of the manufacturing operations.
An audit procedure should include:
Formal procedures should start with an objective.
In this case, the audit objective was discussed in the opening
paragraphs of this chapter -- to monitor the quality system and
take any needed corrective action. The audit scope should include
all functions that impact on whether devices will meet specifications.
These functions include personnel training, facilities, environment,
design controls, device master and history records, equipment
calibration, suppliers, label control, process controls and validation,
complaint files, data feedback, preparation for FDA GMP inspections,
etc. Manufacturers that have a total quality system composed of
a design quality system and a manufacturing quality system should
audit the entire system; otherwise, it is no longer a total quality
system! Audits should cover all buildings and operations as necessary
to make certain that the desired or required quality system is
properly implemented.
The quality system audit required by the
QS regulation is not intended to be a product audit. However,
the adequacy of procedures used to determine product acceptability
should be audited periodically. Product audits and review of the
device master record are desirable as independent evaluations
of product quality to determine the product's fitness for use
and conformance to specification and, these may be acceptable
in satisfying most quality audit requirements when product and
process are very simple and the operation is small. As products
and processes become more complex, evidence from inspection and
testing of products no longer provides full assurance that the
manufacturing system will consistently produce quality products.
Instead, full quality system audits are required to make certain
that:
Manufacturers are responsible for deciding
the frequency of audits. The frequency should depend upon previous
audit findings, any indications of problems, and known stability
of the manufacturing process. If an audit reveals no problems,
the audit intervals could be lengthened -- if problems are identified,
audits may need to be conducted more often. Audits are usually
conducted every 6 to 12 months, but should not exceed 12 months.
Some companies split their audit into parts, and perform one or
more parts per month or quarter, or audit one or more operations
per month or quarter. This approach is valuable because it tends
to direct attention toward problems that can be resolved within
reasonable time limits and existing budgets. However, such segmented
audits may fail to identify company-wide problems. Thus, reviewers
of segmented audit reports should look for indications of company-wide
problems.
The QS regulation (820.22) requires that
quality system audits be conducted by individuals not having direct
responsibility for the matters being audited. This requirement
may be satisfied by an audit team consisting of persons representing
product development and manufacturing. Then, when the product
development area is being audited, the manufacturing persons should
have the lead responsibility and vice versa. For any element of
the quality system being audited, at least one member of the team
should not have direct responsibility for the element being audited.
Management should designate one member as the team leader for
a given audit in order to support consistency, timeliness, completeness,
and uniform response. Of course, a consultant, corporate, or other
independent auditor may be used.
The requirement for an independent audit
should generally be met; however, if a very small manufacturer,
particularly one in which everyone is directly involved in daily
design and production activities, concludes that independent audits
would be unduly burdensome or impractical, the requirement for
independence may be waived. However, if FDA finds, as a result
of inspection or other means, this waiver has compromised the
quality system, FDA may require an independent audit, increase
the frequency of FDA GMP inspections, or take other appropriate
regulatory action.
Individual(s) responsible for conducting
audits should be sufficiently trained and experienced to detect
variations and problems in the quality system [820.20(b), 820.22].
An auditor is expected to objectively compare existing employee
training, design controls, manufacturing processes, facilities,
environmental control, records, test/inspection activities, label
control systems, feedback, etc., against what they should be.
To do this, the individual(s) should have a working knowledge
of:
As with any GMP training, a record shall
be maintained of the audit training given each employee.
Because the quality system requires a written
audit report, auditors should have sufficient writing skills to
effectively communicate findings and recommendations. The effectiveness
of the audit begins with the planning. The manufacturer should
start by defining the purpose and scope of their audit keeping
in mind their quality systems requirements. An audit team leader
and the other members of this team should be identified early
in the planning process. The members of this team should possess
skill and knowledge of quality system principles. Preparing an
audit checklist will enable the team to properly cover the quality
system requirements. Review of previous audits and their resulting
reports is an excellent way for the audit team to correctly evaluate
their quality system audit program. The background preparation
should also include becoming familiar with company policies, operations,
and products. The audit team should notify the parties they will
audit and also hold a pre-audit conference among the audit team
members to clarify exactly what the audit will include and what
the objective(s) of their audit will be. Thus, preparing for an
audit should include elements such as:
Each manufacturer shall determine the criteria
to be used for conducting the audit. In general, medium to large
manufacturers will need extensive documentation outlining the
areas to be audited and the acceptable criteria for each of these
areas. The GMP requirements are a baseline for the evaluation
criteria; however, because the QS regulation is broad, each manufacturer
shall tailor the criteria to the design and manufacturing operations
they are actually performing. Small manufacturers may need only
minimal documentation, and this may consist of an audit checklist
with appropriate ancillary instructions to assure that all aspects
of the quality system are covered.
An audit checklist may be a series of questions,
phrases, trigger words, or any combination of these that will
prompt auditors to cover the entire quality system. Checklists
should cover requirements of the QS regulation applicable to company
products, operations, and other areas company management has decided
are included in their total quality system. If operations or devices
change, evaluation criteria and checklists shall be appropriately
updated.
A quality system audit program that has
been established in accordance with the QS regulation and implemented
in sufficient depth can detect undesirable variations and trends
in operating procedures. Management awareness of these undesirable
variations should lead to corrections and help prevent the design
and production of unsafe, unreliable, or ineffective devices.
The QS regulation requires follow-up corrective
action, including re-audit. When indicated, audit results shall
be given to individuals responsible for each of the operations
audited, especially if deficiencies are found. Audit results shall
be reviewed by all key management personnel, especially those
responsible for the matters audited.
An audit should never be used as a disciplinary
tool. This use will lead to ineffective audits because employees
may become reluctant to reveal any possible problems for fear
of retribution.
Under 704(e) of the FD and C Act FDA has
authority to review and copy all records required by the QS regulation;
however, FDA has elected not to review audit reports. The exception
[820.180(c)] to FDA's policy of not seeking access to reports
of audits of quality systems is that FDA may seek production of
these reports in litigation under applicable procedural rules,
along with other confidential documents. Thus, a copy of the current
audit report should be maintained by the manufacturer. FDA policy
was established because the agency does not wish to prejudice
audits by having auditors concerned that their comments will be
reviewed by FDA investigators. Although FDA investigators do not
have routine access to audit reports, they can request manufacturers
to certify that audits have been conducted and the results documented;
however, investigators do not routinely request certification.
If requested, an employee in management with executive responsibility
should certify, in writing, that the manufacturer has complied
with the audit requirements of the QS regulation.
Investigators usually will ask questions
regarding the audit report such as:
If investigators suspect audits are not
being conducted, questions to determine consistency in answers
may be addressed to those individuals who should have reviewed
these reports. FDA investigators will routinely review audit procedures
and audit checklists.
Two examples of audit procedures with checklists
are included in this chapter. These may be modified to match individual
operations as appropriate or used as guidances.
In response to requests by small manufacturers,
DSMA developed this procedure as an example of a minimum procedure
for quality system audits. Following the procedure are comments
to aid small manufacturers in completing the procedure and developing
a checklist that should be used with it.
No details are given for the format of the
audit "report" because the format generally is not important
for the small manufacturer -- employees of small manufacturers
communicate daily with each other. In fact, the "report"
may be the list of findings neatly noted on the audit checklist.
As noted in the procedure, however, summaries of the audit findings
and corrective actions are recommended. The format Who, When,
etc. discussed in the text, was used to develop this example procedure.
The first three items in this format are reflected in item 1 of
the example and the remaining three are reflected in items 2,
3 and 4 of the example. The scope encompasses the entire quality
system.
Quality System Audit Procedure is an audit
procedure that can be used by a medium to large manufacturer.
In comparison to the small company, there are more people on the
audit team, more audits per year, and the reports are distributed
to more managers, some of which may be at corporate headquarters.
Therefore, this procedure contains more details than the one suggested
for the small company. For example, the procedure dictates the
format of the audit report for the benefit of the managers, who
may review reports for many different operations per year.
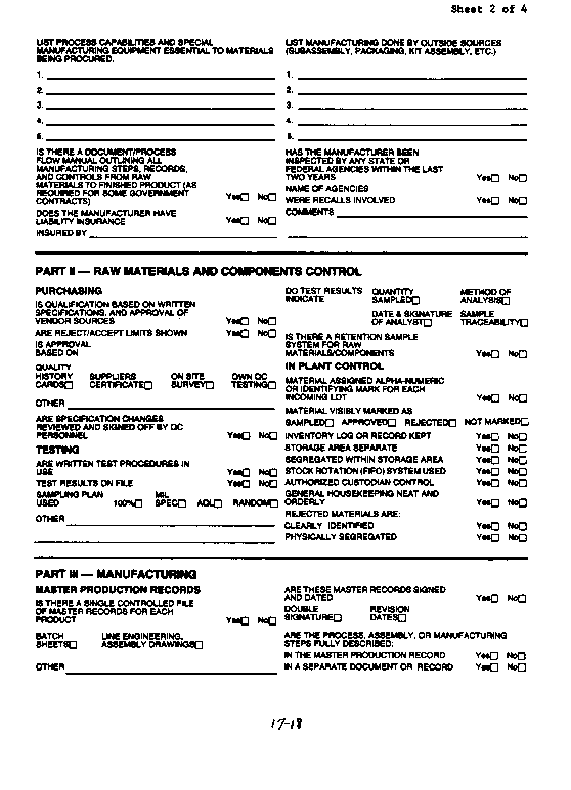
The vendor survey form is applicable to
a vendor or contractor or may be modified and used as an internal
audit checklist. This survey form is divided into areas of concern
such as raw material and component control, manufacturing, quality
control/assurance, etc. Also, it is a more conventional checklist
with places to check off answers to the questions. This form includes
a header with space for manufacturer name, address, date prepared,
etc., and general information about the manufacturer such as,
annual sales, years in business, other plant locations, etc. Your
manufacturers can look at these two styles of checklists and decide
to use one or the other, a combination of both, or a totally different
format.
Sheet 1 of 6
Title: POLICY/PROCEDURE FOR QUALITY SYSTEM
AUDIT No. _____ Rev. ______
Approved by_____________________________________Date______________________________
1. A general audit of the entire quality
system shall be performed by________________________ every_______
months (an audit team may be used).
2. The latest company approved audit checklist
(number) _________shall be used. The audit checklist shall be
updated as required and approved by __________________________to
reflect our current quality system needs.
3. The completed checklist and audit results
summary report shall be reviewed with the following managers,
as appropriate, who are responsible for the matters audited:
____________________, _______________________________________and____________________________________
. Minutes of the review meeting, including a list of attendees
and desired corrective actions, shall be taken, distributed and
filed by__________________________________ . This same procedure
shall be used when reviewing the findings of GMP inspections by
FDA investigators. (820.20)
4. Corrective actions shall be taken by
all affected persons as discussed in the review meeting. ____________________________
will coordinate the corrective actions, re-audits, and keep management
informed. A summary report of the status of the corrective actions,
as determined by a re-audit of the affected areas or other appropriate
means, will be written by _________________________ and filed
with the original audit report. The status report shall be updated
at least bimonthly if there are any uncompleted corrective actions.
A. "Rev."
is the revision level of the latest company approved procedure.
B. The above blanks should be completed
with employee position titles and, if desired, employee names.
C. An audit checklist may be a detailed
series of questions, phrases, trigger words or any combination
of these to assure that the auditor covers the entire quality
system. The checklist should cover the requirements of the QS
regulation applicable to each company's products and operations
plus other areas that company management had decided are included
in the total quality system. A suggested way to develop a question-type
checklist is to refer to the table of contents of the QS regulation
and the chapters of this manual. Then generate questions for each
topic as applicable to specific products and operations of the
manufacturer. If operations or devices change, the checklist should
be updated. A small portion of a quality audit checklist follows.
Sheet 2 of 6
A few cites are given. There are more. For
example most of the QS regulation applies to labeling -- not just
820.120.
SPECIFICATION CONTROLS (820.30,
.40, and .181) YES NO COMMENTS
1. Is an adequate system in place to control
all engineering drawings, specifications
and other related documentation?
2. Does the system require adequate review,
and approval of all new documentation and
changes to documents?
3. Does the system require controlled,
timely distribution of new specifications
and specification changes?
4. Are procedures provided and adequately
implemented to assure collection of obsolete
documentation?
5. Are all specifications used in production
approved, dated, and current?
6. Etc.
Sheet 3 of 6
PROCESS CONTROLS
(820.70, .40, .60 and .80, .86) YES NO COMMENTS
1. Are current, approved process specifi-
cations/procedures such as work instruction,
etc., used to define each process?
2. Are process changes made according to a
formal change system and documented?
3. Are process changes communicated in a
timely manner?
4. Do process specifications and procedures
properly reflect the work to be accomplished?
5. Are adequate acceptance and rejection criteria
provided for the output of each process?
6. Are in-process and rejected items adequately
identified and/or segregated to prevent mix-ups?
7. Etc.
Sheet 4 of 6
PERSONNEL 820.20(b)(2),
820.25, and 820.70(d) YES NO COMMENTS
1. Are there sufficient personnel having the
necessary education, background, training,
and experience to assure that all design and
manufacturing operations are correctly performed?
2. Are training programs conducted and documented?
Is the program proactive? Do design personnel
have basic training in safety, use of standards,
labeling, and applicable regulations?
3. Are all employees made aware of device
defects which may occur from the improper
performance of their specific jobs?
4. Are all employees including salespersons
made aware that they must report all
complaints received from any source to
the company complaint department?
5. Are quality system personnel made aware of defects
and errors likely to be encountered as
part of their individual quality system function?
6. Are verification/validation personnel made aware
of design errors that may be found?
7. Are personnel in contact with the device or its
environment appropriately:
a. clean?
b. healthy?
c. attired?
8. Etc.
Sheet 5 of 6
QUALITY ASSURANCE FUNCTIONS YES NO COMMENTS
1. Does the quality assurance unit or qualified
designee do the following?
a. review customer purchase orders
b. approve or reject components
c. approve or reject manufacturing materials
d. approve or reject in-process materials
e. approve or reject packaging materials
f. approve or reject labeling
g. approve or reject finished devices
h. approve or reject devices manufactured by
another company
i. review production records
j. approve or reject devices processed by
another company
k. approve or reject devices packaged by
another company
l. approve or reject devices held under
contract by another company
m. help provide solutions for quality system problems
n. verify implementation of solutions for
quality system problems
o. assure that all quality system checks are appropriate
and adequate
p. assure that all quality system checks
are performed correctly
2. Are periodic audits of the quality system conducted
to verify compliance with quality system program
requirements?
3. Are audits of the quality system program:
a. performed in accordance with written
procedures?
b. conducted by appropriately trained
individuals?
c. conducted by individuals who do not have
direct responsibility for matters being audited?
4. Are audit results:
a. documented in written audit reports?
Sheet 6 of 6
QUALITY ASSURANCE FUNCTIONS YES NO COMMENTS
b. reviewed by management having
responsibility for the matters audited?
5. Does company have a copy of the last
audit report on file?
6. Is follow-up corrective action, including
re-audit of deficient areas, taken when
indicated?
7. Etc.
QUALITY SYSTEM AUDIT PROCEDURE
Sheet 1 of 3
No._______ Rev._____ Approved___________________________________ Date _____________
ECN History____________________________________________________________________
___________________________________________________________________________________
POLICY:
Periodic and planned audits of systems, training documentation
processes, product flow and feedback shall be performed to assure
compliance with regulatory and company requirements for current
Good Manufacturing Practices (quality system).
SCOPE:
All facilities, design activities, manufacturing operations, and
product lines.
PROCEDURAL GUIDE: An
audit of design activities shall be done annually. Routine quality
audits of selected production areas shall be conducted each month.
The entire operation shall be covered during a 12-month cycle.
An area may be audited more than once. An "Action Audit"
for any area or element may be initiated by the Manager of Quality
Assurance at any time if a special problem arises.
The teamwork approach shall be used to identify
and correct deficiencies.
The audit team shall consist of the Senior
Quality Auditor (team leader) plus one or more individuals from
other disciplines who have no direct responsibility for the area
being audited. A team auditing an Operations unit should include
an R&D representative. A team auditing a quality systems unit
should include an Operations representative. An audit of design
activities shall include a representative from both the regulatory
and the manufacturing divisions.
The Manager of Quality Assurance selects
the team member in consultation with the Department Managers.
A. AUDIT PREPARATION - The
Quality Auditor (team leader) reviews applicable change control
records subsequent to a design transfer, any FDA clearance delay
information, recall records, standard manufacturing procedures,
device histories, complaint history, device labels and inserts,
previous audits with results, follow-up audits, plus any other
document relative to the audit.
B. AUDIT INITIATION - The
Quality Auditor prepares/updates an audit checklist for systematic
examination of the area to be audited, informs the Manager of
the department being audited at the start of the audit, and reviews
observations with the Department Manager.
C. AUDIT ANALYSIS -
The Quality Auditor reviews the data gathered, verifies important
details, and writes an audit report according to the format delineated
in the attached audit report outline.
Sheet 2 of 3
D. ISSUANCE OF AUDIT REPORT - The
Quality Auditor issues the written audit report to the President
and Department managers within three working days following completion
of the audit. If conditions are critical, the Director of Quality
Assurance shall verbally brief appropriate staff members within
12 hours following audit completion. Audit reports shall be stamped
"Confidential".
E. CORRECTIVE ACTION - The
appropriate Management staff member shall be responsible for developing
a schedule for correcting deficiencies cited in the audit report
and submitting same within five working days to the Quality Assurance
Manager. Included in the correction schedule shall be the responsible
individual, and the date when corrective action will be completed.
The Manager of Quality Assurance shall act as arbiter, if necessary,
to judge validity of the deficiency, responsible individual, and
reasonable date to complete the corrective action.
F. AUDIT FOLLOW-UP -
The Quality Auditor maintains a log listing deficiencies, responsible
individual, target date for corrective action, and actual date
of correction. If the same deficiency occurs on a second follow-up
audit, the President shall be notified in writing by the Quality
Assurance Manager.
G. LOG OF AUDITS AND FOLLOW-UP AUDITS
- The master log shall be maintained
by the Senior Quality Auditor. The audit log file shall include
a copy of current audits, list of areas to be audited during the
12-month period, and list of areas audited to date (i.e., part
of the Master Log).
H. REPORT NUMBERS - Audit
numbers shall be composed of the date followed by the sequential
number of the audit being reported (e.g., 98–4 for the 4th
audit during 1998).
AUDIT REPORT COVER DATA
Area Audited ____________________________
Audit No._____________ Date: ____________
Audit Team members________________________________________________________________
_________________________________________________________________________________
Sr. Auditor's Signature: ___________________________________________________
(Team Leader)
REPORT OUTLINE
1. PURPOSE AND AREA DESCRIPTION - Describe
initiating factors for the audit, limitations of audit, and area
being audited.
2. MAJOR FACTS - Summarize
for management review the most undesirable conditions and practices
in order of their relative importance.
Sheet 3 of 3
3. OBSERVATIONS AND FACTUAL DETAILS -
Give a detailed account of the
current practices and the deficiencies listed in four below.
4. DEFICIENCIES -
List deficiencies in procedures, standards, documentation, safety,
etc., along with identity of relevant regulation, SMP, SOP, etc.
5. FOLLOW-UP - State
plans for follow-up review to establish individual responsibilities
and completion dates.
(Download Vendor Survey Form in Printable PDF form)



Updated January 1, 1997
![]()
CDRH Home Page | CDRH A-Z Index | Contact CDRH | Accessibility | Disclaimer
FDA Home Page | Search FDA Site | FDA A-Z Index | Contact FDA | HHS Home Page
Center for Devices and Radiological Health / CDRH



