
INTRODUCTION
QUALITY SYSTEM PRACTICES
Design Controls
Component Selection
Labeling Content
Process Quality
Management Responsibility
Formal and Documented Quality System
Approval of Product
Quality Acceptance Activities
Quality System Audits
Employee Training
QUALITY SYSTEM MAINTENANCE
MEDICAL DEVICE REPORTING
The Quality System (QS) regulation requires that each manufacturer
shall establish and maintain a quality system that is appropriate
for the specific medical device(s) designed or manufactured (820.5
and 820.20). The GMP requirements are harmonized with the International
Organization for Standards (ISO) 9001:1994 and ISO DIS 13485.
The quality system should be an integrated effort -- a total systems
approach, to satisfy the particular safety and performance needs
of a specific manufacturer, product, and user-market. The quality
assurance (QA) activities do not simply consist of inspection
and testing spot solutions or "fire-fighting," no matter
what the product is or how small the manufacturer. In all cases,
quality should be considered at the earliest stages in every significant
area that has an effect on the quality, safety, and effectiveness
of the device. These areas include product development, design
verification and validation, component and/or supplier selection,
documentation, development of labeling, design transfer, process
development and validation, pilot production, routine manufacturing,
test/inspection, device history record evaluation, distribution,
service or repair, and complaints. Complaints and, of course,
favorable comments constitute customer feedback that may result
in improvements in the device, labeling, packaging or quality
system.
Most important of all is management commitment. Management and
employees should have the correct attitude if their quality system
program is to be effective. Quality consciousness should be developed
in every employee. Each person should be made aware of the importance
of his or her individual contributions in the overall effort to
achieve an acceptable level of quality.
After a quality system is in place and checked, it should not
be allowed to stagnate -- it should continue to be dynamic. The
system remains dynamic through continuous feedback, "big-picture"
monitoring by system audits, management review, and corrective
and preventive action. Sufficient personnel with necessary education,
background, and experience should be in all departments to ensure
that quality system activities are properly and adequately performed.
The result is an organization that is operating in a known state-of-control
for the device design, process design, manufacturing processes,
and records. A properly functioning quality system results in
increased safety and effectiveness of the device, reduced liability
exposure, reduced regulatory exposure, increased customer satisfaction,
less scrap, lower costs, much less confusion, higher employee
morale, and, as a result, higher profits.
There are several QA systems in common use, including quality
control, good manufacturing practices, product design assurance,
the ISO 9000 series of international QA standards, and total quality
assurance. Quality control is a minimal system which emphasizes
test and inspection. The QS regulation is a government mandated
QA system for medical device manufacturers. It emphasizes device,
labeling, packaging and process design and all aspects of production:
facilities, equipment, design development, design and production
documentation, correct design transfer, production control, production
records and feedback. Total quality assurance is a system which
emphasizes that: all employees and suppliers are responsible for
their activities; design requirements are established and met;
process requirements are established and met; all production activities
are controlled; finished product specifications are met; and feedback
results in appropriate corrections.
Product design assurance is a QA system which assures that customer
needs are determined, and that product design requirements are
established and met. The ISO 9000 series of QA standards ranges
from basic quality control to very significant design and production
systems.
ISO 9001 is the most comprehensive because it covers design, production,
servicing and corrective/preventive activities. The FDA GMP requirements
are slightly more extensive because they include extensive coverage
of labeling, and complaint handling.
An ideal system for quality assurance is discussed in order to
explain the concept of a system. An ideal QA system is composed
of an organization that executes a QA program according to documented
policy and specifications in order to achieve stated objectives
as shown in Figure 2.1.
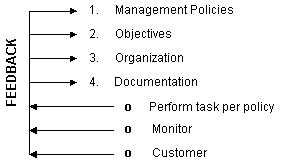
The written policies and objectives are set by management and
are influenced by outside factors such as customer requirements,
standards, and regulations. For example, the customer requirements
and needs and resulting device specifications should be known
to be correct, as these are based on market research, technical
and medical considerations, consensus standards, review of existing
devices, environmental and compatibility considerations, and design
review. The objectives are to produce safe and effective devices
at a profit. Ideally, the quality system includes everyone in
the company as everyone is fully committed to the quality system
program. In addition, however, quality assurance departments such
as design QA and production QA are established to help achieve
specific objectives. Tasks to be performed to meet these objectives
are described in procedures and other documents.
Documentation for a quality system is composed of: product-specific technical documentation such as engineering drawings, component purchase specifications, procedures for manufacturing processes and testing; labels, etc.; and general quality system documentation, such as standard operating procedures (SOP's) for employee training, audits, etc., that are applicable for all products. All activities and product quality are monitored; and any deviations from device and process specifications and company policies are fed back into the system where the deviations are corrected. Likewise, complaint and service information are processed and fed back for appropriate corrections. If the required activities including the feedback are performed, the quality system is self correcting and, thus, the manufacturer is operating in a state-of-control. FDA requires manufacturers of medical devices to operate in a state-of-control.
An adequate and properly implemented quality system such as the
one required by the QS regulation or ISO 9001, because of its
broad scope, has a high likelihood of preventing the design, manufacture,
and shipment of defective products. Basic quality controls such
as inspection and testing, are important parts of a quality system
because they provide information that should be fed back into
the program where action can be taken to correct root causes of
quality problems. Identifying and solving quality problems is
a core requirement of the QS regulation. This approach is in contrast
to merely applying superficial corrections by pass/fail quality-control
inspection including rework of finished product or in-process
assemblies.
Feedback is necessary to verify the adequacy of the design, manufacturing
processes, and the controls used. It also helps trigger corrective
action to solve root causes of quality problems rather than just
performing rework.
Each manufacturer is required by regulation to establish and maintain
design control procedures for any class III or class II device,
and a selected group of class I devices. The class I devices subject
to design controls are devices automated with computer software
and the following specific devices:
| SECTION | DEVICE |
| 868.6810 | Catheter, Tracheobronchial Suction |
| 878.4460 | Glove, Surgeon's |
| 880.6760 | Restraint, Protective |
| 892.5650 | System, Applicator, Radionuclide, Manual |
| 892.5740 | Source, Radionuclide Teletherapy |
Because the intrinsic quality level of devices and processes is
established during the design phase, the quality system program
should include this phase if the program is to assure overall
quality, meet customer requirements, meet company quality claims,
and comply with the intent of the FD&C Act. The terms "product
assurance" and "design QA" are often used to identify
the quality system activities related to product design. The QS
regulation uses the term "design controls." A product
assurance system or design QA system combined with a production
QA system constitutes a total quality system.
Quality system, production, regulatory, and other appropriate
personnel should participate in the review, evaluation, and documentation
of the components, device, and process design. It is from data
established during this preproduction phase that all other activities
derive such as, purchasing, processing, and testing. Development
and validation data are also useful in cases of regulatory or
product liability actions to show that the design and manufacturing
processes were well conceived and properly validated, reviewed,
and documented.
Total quality systems extend from customer requirements through
development and production to customer use and feedback. Thus
total quality systems encompass the medical device law and regulations,
particularly the QS regulation. The FD&C Act, and its implementing
regulations such as those for Labeling, Premarket Notification,
Investigational Device Exemptions (IDE), Premarket Approval (PMA),
and GMP requirements impact the quality of devices at various
times during the design product life-cycle. The IDE, PMA, 510(k),
labeling and QS regulation with their preproduction and production
requirements constitute a total quality system. For example, Section
501(c) of the Act states that a product is adulterated if it does
not have a quality equal to the quality stated or implied by the
product labeling. Analysis of device recall problem data by FDA
has shown that such problems are divided almost equally between
design and production. Thus, a production quality assurance program
is not sufficient to produce safe and effective devices -- design
shall also be covered. A design quality assurance system is required
by the QS regulation.
Two other reasons for having a total quality system are 21 CFR
Part 803, Medical Device Reporting (MDR), and product liability.
MDR requires manufacturers of medical devices to report to FDA
certain adverse events that they receive from any source. Product
liability actions are often the result of poor design, labeling,
and manufacturing. Reporting and liability exposure are reduced
by using a total quality system.
Intrinsic or desired quality is established by the design specifications
for the product, its components, and the manufacturing processes.
Complying with the QS regulation assures that the manufacturing
processes can consistently achieve desired levels of quality and
that the finished device meets its device master record specifications.
This result is a significant quality step. However, if the device
as designed is of poor quality, the GMP production controls will
only assure that a poor quality device is manufactured. Thus,
the QS regulation requires an overall quality system program,
which embraces evaluation of customer needs; product design; verification
and validation; labeling development and control; all manufacturing
and control activities; and customer feedback.
Component and raw material specifications developed during the
design phase should be well conceived and adequate for their intended
purpose. New components or components for an unusual application
need to be verified (qualified) for the intended use. In some
cases, where large quantities of components or raw materials are
involved, the specifications should include valid and well understood
methods of sampling and acceptance. These specification and sampling/acceptance
plans should also be accessible and acceptable to suppliers. The
specifications are device master record (DMR) spec document or
the specifications appear in a DMR drawing or procedure.
Manufacturers shall establish and maintain procedures to ensure
their purchased and otherwise received products and services conform
to their specified requirements. The manufacturers shall then
assess their suppliers, contractors, and consultants based on
their ability to meet the established specifications. When possible,
an agreement shall be established to include that the suppliers,
contractors, and consultants will notify the manufacturer of any
changes in the product or service that may affect the quality
of a finished device.
The regulations in 21 CFR Part 801, Labeling; Part 809, In Vitro
Diagnostic Products for Human Use; and Part 812, Investigational
Device Exemptions, are intended to control the content of labeling.
Likewise, 21 CFR Part 807, Premarket Notification; and Part 814,
Premarket Approval and 820.30, Design Controls, help control the
content of labeling by design and premarket submissions. The intent
of these regulations and the FD&C Act is for manufacturers
to have a labeling control program such that their labeling always
complies with the regulations and meets the needs of the users.
By a formal process under a total quality system during the design
phase, clear and concise printed and/or software labeling are
written and reviewed; and the ink substrate and attachment methods
for printed labeling are developed. Such labeling is designed
to meet customer and regulatory requirements. Thereafter, the
procurement, use of the correct label, and the correct attachment
of labels is assured under a manufacturer's quality system elements
for these activities.
Manufacturing methods and processes to be used should be developed,
equipment selected, and processes and methods qualified. For all
significant processes such as welding, molding, lyophilizing,
sterilizing, and packaging/sealing where the output cannot be
fully verified, the qualification should include a full validation
of the processes. The output may not be fully verified for economic,
technical, or practical reasons and thus validation is needed.
Production specifications and methods employed in manufacturing
should result in standard in-process and finished products without
excessive sorting or reprocessing. Inspection and test methods
should be developed that will adequately monitor product characteristics
to make certain these are within the acceptable specifications.
These methods should be developed, evaluated, validated where
necessary, and documented during the product and process development
phase. The methods should be implemented at the beginning of routine
production.
Any adverse effects the manufacturing processes, manufacturing
materials, or equipment may have on device safety and performance
should be identified. Where necessary, procedures have to be developed,
implemented, and monitored to control these characteristics. Quality
system personnel should participate in the timely (i.e., early)
development of special controls, test or inspection methods, or
training programs needed to insure product quality. Acceptance
methods should be developed for accurate measurement of outgoing
product quality.
As set forth by the QS regulation (820.20), one of the most important
responsibilities of management when developing a quality system
is to establish its policy and objectives for, and commitment
to, quality. Management with executive responsibility shall ensure
that the quality policy is understood, implemented, and maintained
at all levels of the organization. This means each manufacturer
shall establish the appropriate responsibility, authority, and
interrelation of all personnel who manage, perform, and assess
work affecting quality, and provide the independence and authority
necessary to perform these tasks. The QS regulation also requires
that each manufacturer shall establish and maintain an adequate
organizational structure to ensure that devices are designed and
produced in accordance with the GMP requirements. To meet these
regulatory requirements, manufacturers are required to provide
adequate resources, including the assignment of trained personnel
for management, performance of work, and assessment activities,
including internal quality audits.
Management with executive responsibility shall appoint a member
of management who will have authority over and responsibility
for:
Thus, the QS regulation requires that management with executive
responsibility shall review the suitability and effectiveness
of the quality system at defined intervals and with sufficient
frequency according to established procedures to ensure that the
quality system satisfies the regulatory requirements and the manufacturer's
established quality policy and objectives. The dates and results
of quality system reviews shall be documented.
The quality assurance personnel should be able to identify system
problems, to recommend and provide solutions, and to verify implementation
of the solutions. Other personnel may also identify and solve
quality problems. The quality system should support such activities
by all personnel. Feedback from quality assessment activities
is necessary to verify the adequacy of the manufacturing process
and the controls used. It also helps trigger corrective action
to solve root causes of quality problems rather than just performing
rework.
Typically, a quality system identifies problems with device quality
through review of verification and validation data, inspection/test
data, analysis of device history and service records, failure
analysis, analysis of complaints, and review of other objective
data. In this regard, reduction in productivity is often an indicator
of quality problems. Low morale and confusion are indicators of
inadequate procedures, and/or training and poor management. Also,
measurement of scrap and rework is an effective method of detecting
quality problems and reducing costs. These are examples of sources
that provide feedback to the quality system.
In conclusion, each manufacturer is required to establish a quality
plan which defines the quality practices, resources, and activities
relevant to the devices that are designed and manufactured. The
manufacturer shall establish how the requirements for quality
will be met [820.20(d)]. Each manufacturer shall establish quality
system procedures and instructions. To facilitate the understanding,
use, review, and updating of the quality system, an outline of
the structure of the documentation used in the quality system
shall be established where appropriate [820.20(e)].
The QS regulation requires that each manufacturer prepare and
implement quality system procedures adequate to assure that a
formally established and documented quality system is implemented
The system should include not only formal documentation, but also
an obvious commitment to quality from top management. There should
be manifest indications that management recognizes the need for
a quality system in order to assure quality products. In many
manufacturers, this commitment is accomplished through means such
as: a management policy; assignment of responsibilities and authorities;
and general statements and actions such as employee training that
define goals of the quality system. This policy is supported by
a number of more detailed quality system documents such as verification
methods, sampling procedures, inspection/test procedures, product
audits, and records indicating that measurement and monitoring
of quality has occurred. The number of documents needed depends
on the size and complexity of the operation and the characteristics
of the product. The QS regulation requires the manufacturer to
maintain various records such as:
Most of these records are discussed in more detail in later chapters.
In each case, the records should be appropriate for the device
and the operation involved. Any changes to device master records
should be made by a formal procedure and be formally approved.
Among other records, the device master record contains manufacturing
procedures and standard operating procedures (SOP's). Some manufacturers
tend to write an excessive number of general SOP's. Manufacturers
should not generate and use procedures that are not needed. Also,
standard operating procedures tend to not match actual operations
because the operations gradually change as the company grows or
as products are added without amending the procedures. Such procedures
may require operations that have no benefit, or require excessive
collection of data, or collection of data that is never used.
Thus, manufacturers need to occasionally flow chart and analyze
their operations to determine, among other things, if the existing
procedures are inadequate, correct, or excessive. Flow-charting
is a tool that directs a detailed audit of an operation. Flow-charting
to analyze operations is an excellent method for improving operations
and the associated quality system activities. At the end of Chapter
10, Purchasing and Acceptance Activities, an example of a flow-chart
is contained in PA1004, Procedure for Receiving and Inspection
of Material, integral page 4 of 9.
The quality system includes procedures for assuring that all products
such as components, packaging, labeling, manufacturing materials,
and finished devices have been approved for use; and that contracted
items and services are suitable [820.50, 820.80]. Likewise, the
quality system shall assure that rejected items are identified
and properly disposed [820.90]. Additionally, the quality system
shall assure that production records are reviewed before the product
is distributed [820.80(d)]. These records are part of the device
history record. Device history records shall be reviewed to verify
that the operations represented have been properly conducted and
that the records are complete.
The quality system shall determine that all tests and inspections
are performed correctly (see 820.80, 820.181, and 820.20). Some
of the methods used to accomplish this are adequate test and inspection
procedures, training of test personnel, quality system audits,
review of quality system records, and product audits. However,
simply instituting a quality system and checking that it is conducted
correctly is not enough to satisfy the QS regulation. The regulation
also requires that the quality system be appropriate and adequate
for the purpose. This determination should be done during final
product development, pilot production, and, of course, whenever
product and/or processes are modified. In cases where conformance
to specifications cannot be adequately measured by in-process
or finished product testing and inspection, the system should
include validation of processes.
The QS regulation requires (820.20) that each manufacturer shall
prepare and implement quality system procedures adequate to assure
that a formally established and documented quality system program
is performed. Many activities are required to fulfill this requirement.
As management performs their assigned routine duties, they should
be aware of the obvious aspects of the quality system. However,
to make sure that all aspects, obvious, hidden or subtle, of the
required program exist and are operating correctly, the QS regulation
requires planned and periodic audits (820.22) of the quality system.
Management with executive responsibility reviews audit reports
as part of their review of the suitability and effectiveness of
the quality system.
QS regulation requires quality awareness training for manufacturing
and quality system personnel [820.25(b)]. Personnel involved in
quality system activities shall be properly trained, both by education
and experience. No matter how effective quality system and production
systems are as concepts, people still play the major role in producing
a quality product. Lack of training -- as reflected in instances
of negligence, poor operating techniques, or inability of employees
to discharge their functions properly -- can lead to defective
products and, sometimes, to regulatory or liability problems.
Management should be diligent in looking for factors that indicate
a need for employee training.
A quality system should include an ongoing formal program for
training and motivating all personnel. All employees should be
made aware that product quality is not solely the responsibility
of management. Quality is the responsibility of every employee
-- any employee can potentially generate a quality problem through
negligence. It is extremely important to understand the following
points with respect to typical quality-related functions.
A medical device manufacturer should NEVER try to operate on the
basis that only the quality system organization has primary and
direct responsibility for the quality of the products. To do so
means that quality problems will not be solved in a timely manner
because attention is directed toward the wrong organization. In
reality, it is part of the responsibility of the quality system
to see that attention is directed toward the correct department
if a quality problem arises.
Where necessary, employees should be certified to perform certain
manufacturing or quality system procedures. Records of training
and/or certification shall be maintained. Personnel performing
quality system functions should:
After the quality system is operational, personnel should continue
to look for problem areas or factors that can have an impact on
product quality. Many factors that can have an impact on product
quality include:
As noted, quality system audits and flow-charting of operations
are excellent methods for determining the detailed status of the
system. Correcting problems or responding to conditions identified
by audits, operational analyses, and customer feedback data can
result in quality system improvements.
FDA has promulgated regulations [803] for manufacturers, distributors,
and initial distributor(s) requiring them to establish and maintain
reports, including the Medical Device Reporting (MDR) reports
for serious injuries, death, or certain other adverse incidents.
If a manufacturer has a quality system as required by the QS regulation,
the frequency of MDR reporting should be minimized.
Updated January 1, 1997
![]()
CDRH Home Page | CDRH A-Z Index | Contact CDRH | Accessibility | Disclaimer
FDA Home Page | Search FDA Site | FDA A-Z Index | Contact FDA | HHS Home Page
Center for Devices and Radiological Health / CDRH



