Los Hechos Acerca de Las Distrofias Musculares Poco Comunes
Las Distrofias Musculares de Emery-Dreifuss, Congénita, Distal Y Oculofaríngea
Queridos Amigos:
 Mike Neufeldt Mike Neufeldt |
Cuando yo tenía como un año, mis padres notaron algo extraño en mi forma de caminar. Después de muchos exámenes, se llegó a la conclusión que yo tenía una enfermedad neuromuscular. Nos tomó muchos años obtener un diagnóstico definitivo de distrofia muscular de Emery-Dreifuss.
Si usted ha descubierto recientemente que tiene una forma poco común de distrofia muscular, podrá entender lo que tuvo que pasar mi familia. Debido a lo poco común de las distrofias musculares de Emery-Dreifuss, congénita, distal y oculofaríngea, es muy importante que usted obtenga toda la información que pueda acerca de su trastorno. Este panfleto es un buen comienzo.
Descubrir que usted o su hijo tiene una forma poco común de DM puede ser aterrador y confuso. Mis padres se preguntaron por qué tenía yo esta enfermedad; en nuestra familia no hay historia de haberla tenido. Pero, como se explica en este panfleto, cada tipo de DM es causado por un defecto genético extremadamente poco común, que a menudo las personas ni siquiera saben que lo tienen. Usted puede estar seguro que su trastorno no fue causado por nada que usted o sus padres hayan hecho y tampoco se le contagió de nadie.
Mi familia tuvo que adaptarse de muchas formas a causa de la DM. Pero tuvimos suerte. Junto con doctores excelentes y compasivos, tuvimos la ayuda de la Asociación de la Distrofia Muscular. Mis padres recibieron de la MDA el apoyo y la información que necesitaban, así como ayuda para obtener aparatos ortopédicos y otros servicios. Yo tuve el honor de servir como el Embajador Nacional de Buena Voluntad de la MDA en 1987-88.
Agradezco a mis padres no haber escondido las cosas y haberme permitido formar parte de las discusiones con los médicos desde una temprana edad. Entender mi enfermedad me ayudó a prepararme para manejar mis cuidados médicos como persona adulta.
Contar con información sobre mi DM también me permitió
disfrutar de una niñez típica, incluyendo deportes, Cub Scouts
y muchos amigos. En la secundaria mantuve buenas calificaciones, era
el estadístico del equipo y tenía un empleo de tiempo parcial.
Me gradué de la universidad con un diploma en comunicaciones y soy un diseñador entrenado de sitios de Internet. Actualmente utilizo una silla de ruedas motorizada y asistencia respiratoria parte del tiempo.
Le cuento esto acerca de mi persona para que pueda darse cuenta que las personas con formas poco comunes de DM pueden llevar vidas satisfactorias y felices. No es fácil vivir con músculos que se debilitan con el tiempo, pero no es necesario dejar que la DM le impida alcanzar una educación, una carrera, una familia, viajes — todo lo que usted quiera.
Las personas con discapacidades tienen más oportunidades que nunca para desarrollar y utilizar sus habilidades. Las leyes federales nos garantizan una educación pública, oportunidades equitativas de empleo y acceso a los lugares públicos. Las computadoras y la tecnología nos ayudan a movilizarnos, escribir, trabajar y conducir vehículos.
"La MDA Está Aquí para Ayudarle", describe los valiosos servicios de la Asociación. La MDA es también el líder mundial en la investigación de enfermedades neuromusculares y sus científicos han hecho muchos descubrimientos emocionantes en los últimos años acerca de todas las formas de DM.
Este folleto le proporcionará los datos básicos acerca de estas cuatro formas de DM y la MDA le ayudará a contestar todas sus preguntas conforme se presenten. A medida que le hace frente a los retos que se avecinan, por favor tenga la seguridad que estamos avanzando rápidamente hacia mejores tratamiento y hacia una cura. Y recuerde, no está solo.
Mike Neufeldt
Milwaukee
Equipo de Trabajo Nacional para Concienciación Pública de la MDA
¿QUE ES LA DISTROFIA MUSCULAR?
Las distrofias musculares son un grupo de enfermedades genéticas que causan debilidad y degeneración muscular, principalmente en los músculos esqueléticos o voluntarios (aquellos que controlamos, como los músculos de los brazos y las piernas). Los cuatro tipos de distrofia muscular (DM) que se describen en este panfleto — distrofia muscular congénita (CMD), distrofia muscular distal (DD), distrofia muscular de Emery-Dreifuss (EDMD) y distrofia muscular oculofaríngea (OPMD) — están entre las menos comunes de las distrofias musculares. Debido a que son menos comunes, pueden ser difíciles de diagnosticar y quedan muchas preguntas sin contestar acerca de sus síntomas y su progresión.
Tanto la distrofia muscular congénita como la distrofia muscular distal (a veces llamada miopatía distal) son grupos de trastornos musculares. La de Emery-Dreifuss y la oculofaríngea parecen ser ambas una forma única de distrofia muscular en lo que se refiere a los síntomas (aunque la EDMD y la OPMD pueden tener más de una causa genética).
La mayoría de personas con distrofia muscular experimentan algún grado de debilidad muscular durante sus vidas, pero cada uno de los cuatro trastornos descritos en este panfleto afecta a diferentes grupos de músculos y puede tener síntomas acompañantes diferentes. Debido a que en las distrofias musculares la debilidad muscular generalmente progresa conforme pasa el tiempo, pueden ser necesarios los cambios en el estilo de vida, los dispositivos de ayuda y la terapia ocupacional para ayudar a la persona a adaptarse a situaciones nuevas.
¿Qué Causa la Distrofia Muscular?
Todas las formas de distrofia muscular son heredadas — es decir, son causadas por mutaciones (cambios) en los genes de una persona. Nuestros genes están compuestos de ADN y residen en nuestros cromosomas. Cada gene contiene la "receta" para una proteína diferente y sus variaciones, y estas proteínas son necesarias para que nuestros cuerpos funcionen correctamente.
Cuando un gene sufre una mutación, puede fabricar una proteína defectuosa, o no fabricar ninguna. Más comúnmente, las proteínas faltantes o defectuosas en los músculos evitan que las células musculares trabajen adecuadamente, conllevando a síntomas de distrofia muscular, incluyendo debilidad y degeneración muscular a medida que pasa el tiempo.
Los músculos están compuestos de manojos de fibras (células). Grupos de proteínas dentro de la célula y a lo largo de las membranas que rodean cada fibra, ayudan a mantener las células musculares funcionando adecuadamente. Cuando falta una de estas proteínas o es inadecuada (porque un gene no la produjo adecuadamente), el resultado puede ser alguna forma de distrofia muscular. La ausencia de diferentes proteínas, o los defectos en ellas, están entre las causas de diferentes tipos de distrofia muscular. |
 |
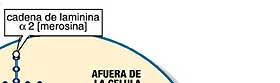
Varias formas de distrofia muscular congénita se derivan de defectos en las proteínas dentro o fuera de la membrana de la célula muscular (fukutina, integrina), o en la matriz extracelular, la cual se adhiere a la membrana (merosina o laminina-alfa-2). |
 |

|
 |
MEMBRANA DE CELULA MUSCULAR
|
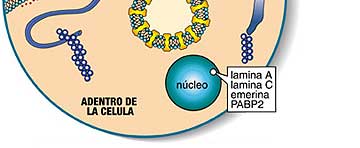
Otra proteína de la membrana, disferlina, está implicada en la distrofia muscular distal. La ausencia de algunas de las funciones de las proteínas en el núcleo de la célula lleva a la distrofia muscular de Emery-Dreifuss (emerina, lamina A, lamina C) o a la distrofia muscular oculofaríngea (PABP2).
(La ausencia de otras proteínas musculares vitales (que no se muestran) lleva a distrofias musculares que no se cubren en este folleto, tal como la DM de Duchenne.) |
|
¿Qué le Sucede a Alguien con Distrofia Muscular?
La mayoría de las formas de distrofia muscular son progresivas y tienden a empeorar con el tiempo. Sin embargo, la edad de inicio y el ritmo de progresión pueden variar mucho de un trastorno a otro. Algunos de estos trastornos, pero no todos, pueden afectar la expectativa de vida. En muchos casos, el conocimiento adelantado permite el tratamiento de los síntomas que tienen más probabilidad de reducir la expectativa de vida.
En la mayoría de los casos de distrofia muscular, la masa muscular en las regiones afectadas puede degenerar en forma visible (reducción del tamaño) y los brazos, las piernas y el torso pueden debilitarse tanto que eventualmente no se pueden mover. Algunas formas de distrofia muscular vienen acompañadas de contracturas, o articulaciones rígidas, y otras son acompañadas de escoliosis, o curvatura de la columna.
Las formas de distrofia muscular que afectan los músculos utilizados para tragar pueden necesitar que se tomen precauciones al comer y beber, de manera que la comida no sea aspirada hacia los pulmones. Aunque la mayoría de distrofias musculares no afectan el cerebro, algunas se presentan con cambios cerebrales que pueden producir discapacidades de aprendizaje que van de leves a severas.
Finalmente, algunas formas de distrofia muscular también afectan el corazón y deben tomarse precauciones especiales para controlar las funciones cardíacas. Cada trastorno tiene sus propias consideraciones especiales.
¿Qué se Puede Hacer para Tratar la MD?
Actualmente no hay cura para ninguna forma de distrofia muscular, pero se cuenta con muchas terapias diseñadas para ayudar a manejar los síntomas comunes de la enfermedad. Por ejemplo, las contracturas pueden aliviarse con terapia física y algunas veces con cirugía para liberar los tendones, mientras que la escoliosis puede responder a dispositivos ortopédicos o cirugía y los problemas del corazón pueden responder a medicamentos o a la implantación de un marcapasos. (Las terapias físicas y ocupacionales pueden iniciarse a través de la escuela de su hijo o por medio de su clínica MDA.)
Muchas personas con estas formas de distrofia muscular viven su vida con mucha plenitud. El director de su clínica MDA le ayudará a planificar la mejor estrategia para hacerle frente a las necesidades específicas suyas o de su hijo.
tapa 
¿ES HEREDITARIO?
Cuando se enteran que tienen un trastorno genético como la distrofia muscular, los confundidos padres a menudo preguntan, "Pero si nadie lo tiene en la familia, ¿cómo puede ser genético?"
La distrofia muscular sí puede ser hereditaria, aun cuando solamente una persona en la familia biológica la tiene. Esto se debe a las formas en que se heredan las enfermedades genéticas.
Cada forma de distrofia muscular sigue uno de tres patrones de herencia: recesivo, dominante y ligado al cromosoma X. En pocas palabras, si una enfermedad es recesiva, se necesitan dos copias del gene defectuoso (una de cada progenitor) para producir la enfermedad. Cada progenitor sería un portador del gene defectuoso, pero normalmente no tendría la enfermedad.
Si una enfermedad es dominante, se necesita solamente una copia del defecto genético para provocar la enfermedad. Cualquiera que tenga el defecto genético tendrá los síntomas de la enfermedad y puede heredar el trastorno a los hijos. Si una enfermedad está X-ligada, se hereda de la madre al hijo varón, mientras que las hijas pueden ser portadoras pero generalmente no tienen la enfermedad.
Muchas veces la DM parece haberse presentado "de sorpresa", pero en realidad, uno o ambos progenitores pueden ser portadores, llevando en silencio la mutación genética (un defecto en el gene).
En raras ocasiones, la distrofia muscular puede de hecho ocurrir "de sorpresa", cuando una mutación nueva aparece con la concepción de un bebé, aunque ninguno de los progenitores sea portador de la falla genética. Estas se conocen como mutaciones espontáneas y, después de que ocurren, pueden ser transmitidas a la siguiente generación.
El riesgo de transmitir una forma de distrofia muscular a sus hijos depende de muchas circunstancias, incluyendo exactamente qué clase de DM ha sido diagnosticada. Una buena manera de averiguar más acerca de estos riesgos es hablar con el doctor de su clínica MDA, o pedir una cita con el consejero genético en la clínica MDA. También puede leer el panfleto de la MDA "La Genética y las Enfermedades Neuromusculares".
tapa
DISTROFIA MUSCULAR CONGENITA
¿Qué Causa la CMD? | ¿Cuáles Son los Tipos de CMD? | CMD de Merosina Negativa | CMD de Merosina Positiva
CMDs con Trastornos de Migración Neuronal | Problemas y Soluciones en las Distrofias Musculares Congénitas

El término distrofia muscular congénita (CMD) es actualmente el nombre dado a un grupo de distrofias musculares que tienen en común que la debilidad muscular comienza en la infancia o en la niñez muy temprana (típicamente antes de los 2 años de edad). Las enfermedades congénitas son aquellas cuyos síntomas se presentan al nacer o poco tiempo después.
Un diagnóstico de CMD puede ser confuso porque, durante muchos años, el término se utilizaba como un nombre "genérico" para describir condiciones que se parecían mucho a otras distrofias musculares, pero que comenzaban mucho antes o seguían diferentes patrones de herencia. En años recientes, los doctores han acordado que hay varias categorías diferentes de CMD "verdaderas", causadas por mutaciones genéticas específicas y que son distintas de otras distrofias musculares. Es posible que algunas personas que han sido diagnosticadas con CMD hace muchos años, en realidad tengan alguna otra forma conocida de distrofia muscular con un inicio excepcionalmente temprano.
Aunque los niños con CMD pueden tener síntomas aso-ciados, grados de severidad y tasas de progresión diferentes, la mayoría muestran alguna debilidad muscular progresiva. Esta debilidad, que usualmente se identifica inicialmente como hipotonía, o falta de tono muscular, puede hacer que un infante parezca "flojo". Más adelante, los infantes y niños que empiezan a andar pueden retrasarse en llegar a las marcas motoras, tales como darse vuelta, sentarse o caminar, o pueden no llegar a alcanzar algunas marcas del todo.
Algunas de las formas menos comunes de CMD también llegan acompañadas de discapacidades de aprendizaje significativas o de retraso mental.
¿Qué Causa la CMD?

Un niño con DM congénita |
Las CMDs son ocasionadas por defectos genéticos que afectan a proteínas musculares importantes. La mayoría de las formas de CMD son heredadas en un patrón autosomal recesivo, pero al menos una forma parece seguir un patrón hereditario dominante. (Para más información sobre patrones hereditarios, ver "¿Es Hereditario?".)
No se sabe por qué las CMDs causan debilidad muscular más temprano que otros tipos de distrofia muscular. Una posibilidad es que las proteínas musculares afectadas en la CMD son necesarias en una etapa temprana del desarrollo de los músculos de un infante, mientras que las proteínas musculares vinculadas con otras distrofias musculares no pasan a ser importantes hasta que los músculos comienzan a ser más utilizados a medida que el niño crece.
Es importante hacer ver que sólo porque la debilidad muscular comienza más temprano en la CMD, la CMD no es automáticamente más severa que otras formas de distrofia muscular. El grado y la tasa de progresión de la debilidad muscular varía con las diferentes formas de CM y de un niño a otro.
¿Cuáles Son los Tipos de CMD?
La CMD no es una condición única. Investigaciones recientes demuestran que la CMD puede dividirse en tres grupos principales: trastornos de merosina negativa, de merosina positiva y de migración neuronal (ver tabla.)
La merosina es una proteína que se encuentra en la delgada capa de tejido conectivo que rodea y sostiene cada fibra muscular. Las personas con CMD, con o sin anormalidades de merosina, tienen diferentes grados de debilidad muscular, y en algunos casos se presenta una discapacidad de aprendizaje acompañante. En los trastornos de migración neuronal (de las células nerviosas), las personas tienen retraso mental severo y enfermedades neurológicas que opacan el trastorno muscular.
| Clasificación de Distrofias Musculares Congénitas (CMDs) |
| Enfermedad |
Cromosoma/Gene |
Patrón Hereditario |
CMD de merosina negativa
(completa o parcial) |
merosina |
autosomal recesivo |
Merosina positiva (MP)
CMD pura
Enfermedad de columna rígida
Enfermedad de Ullrich
Deficiencia de integrina alfa7 |
desconocido
SEPN1
COL6A2
ITGA7 |
autosomal recesivo
autosomal recesivo
autosomal recesivo
autosomal recesivo |
Trastornos de migración neuronal
CMD de Fukuyama (FCMD)
Enfermedad de músculo-ojo-cerebro (MEB)
Síndrome de Walker-Warburg (WWS) |
fukutina
cromosoma 1
desconocido |
autosomal recesivo
autosomal recesivo
autosomal recesivo |
CMD de Merosina Negativa
Los niños con CMD de merosina negativa carecen de toda o parte de la proteína muscular merosina (también conocida como laminina alfa 2). El grado de debilidad muscular puede variar desde severo (nunca llegan a caminar) hasta leve (caminan entre los 2 y 3 años de edad) y depende de cuánta proteína merosina está produciendo el niño.
Esta forma de CMD progresa muy lentamente o, en algunos casos, no progresa para nada. Los problemas especiales incluyen las contracturas, dificultad para respirar y convulsiones (en el 20 por ciento de los casos). La inteligencia es generalmente normal, pero se han documentado discapacidades de aprendizaje.
Una característica de diagnóstico distintiva de este tipo de CMD se encuentra por medio de imágenes de resonancia magnética (MRI). Estas imágenes del cerebro muestran los cambios en la materia blanca, que consiste de fibras nerviosas que transportan mensajes desde el cerebro hasta la médula espinal. A pesar de la apariencia en el MRI, aquellos con CMD de merosina negativa tienen pocas señales de daño cerebral en la vida diaria.
CMD de Merosina Positiva
En este grupo de CMDs, las biopsias musculares muestran la presencia de merosina. Pero en algunas formas de la enfermedad hacen falta otras proteínas.
CMD pura
El cuadro clínico de la CMD pura es similar al de la CMD de merosina negativa. La debilidad muscular puede ser de leve a moderada y las complicaciones pueden incluir contracturas de las extremidades. El cerebro se presenta normal y la inteligencia es normal.
CMD con síndrome de columna rígida (RSMD1)

La CMD puede causar contracturas en las muñecas, los tobillos y otras articulaciones. |
Esta forma de CMD se caracteriza por su inicio antes del primer año de edad, con debilidad marcada en el cuello y poco control de la cabeza. Después de alguna mejora inicial, los niños desarrollan gradualmente (entre los 3 y los 7 años de edad) tiesura o rigidez de la columna vertebral. En menor grado, se ven contracturas de los músculos de las extremidades.
Al llegar a la adolescencia se ven afectados los músculos que operan los pulmones, mientras que se ve menos afectada la fuerza de los músculos de las extremidades. La función intelectual es normal.
El gene defectuoso que lleva a la RSMD1 está en el cromosoma 1 y produce una proteína llamada SEPN1 o selenoproteína. La función de esta proteína está siendo investigada, pero algunos estudios sugieren que protege al músculo contra el daño de los radicales libres.
Enfermedad de Ullrich
Las características clínicas de la enfermedad de Ullrich incluyen hipotonía (pérdida del tono muscular) y contracturas múltiples de las articulaciones al nacer, con rigidez de la columna vertebral y marcada hiperlaxidad distal (articulaciones flojas) de las manos y los pies. El curso es lentamente progresivo, causando debilidad y degeneración muscular. La inteligencia es normal. La mayoría de las personas con la enfermedad de Ullrich tienen fallos respiratorios severos en la primera década de su vida.
La enfermedad de Ullrich se hereda como un trastorno autosomal recesivo. El gene que, cuando es defectuoso, causa esta enfermedad, produce una proteína llamada colágeno que también ayuda a sostener las fibras musculares.
CMD con deficiencia de integrina
Esta forma de CMD de herencia recesiva es causada por un gene responsable de producir una proteína llamada integrina alfa7. Esta proteína tiene interacción con la merosina y también rodea y sostiene cada fibra muscular. Los niños con deficiencia de integrina tienen hipotonía y debilidad en la infancia temprana, asociada con marcas de desarrollo atrasadas. Los niños generalmente no caminan hasta los 2 a 3 años de edad.
CMDs con Trastornos de Migración Neuronal
CMD de Fukuyama (FCMD)
La terapia física es importante para mantener el rango de movimientos y reducir las contracturas. |
La distrofia muscular congénita de Fukuyama se presenta casi exclusivamente en personas de ascendencia japonesa. El trastorno se ha vinculado a un gene llamado fukutina y se piensa que la mutación más común comenzó hace muchos años con un sólo antepasado japonés.
La debilidad muscular en la FCMD varía desde severa hasta leve y las personas con los casos más leves pueden caminar con ayuda. Anormalidades extensas del cerebro están normalmente acompañadas de retraso mental severo, epilepsia, pérdida de la vista y disminución de la expectativa de vida (unos 11 a 16 años de edad).
Enfermedad de músculo-ojo-cerebro (MEB)
Esta forma muy poco común de CMD, descrita por primera vez en Finlandia, comparte características con la FCMD. Generalmente es más leve, con supervivencias que varían desde la niñez temprana hasta los 70 años de edad. La acompañan marcas motoras atrasadas, retraso mental severo y problemas de la vista. El gene de este trastorno no se conoce aún, pero se le ha vinculado con una región específica del cromosoma 1.
Síndrome de Walker-Warburg (WWS)
Esta forma muy poco común de CMD es similar a la MEB, pero más severa. Las personas con este trastorno tienen hipotonía y convulsiones. Se encuentran retraso mental severo y múltiples problemas de la vista. El trastorno es generalmente mortal en la infancia.
Problemas y Soluciones en las Distrofias Musculares Congénitas
Contracturas: Las articulaciones rígidas o "congeladas" (contracturas) pueden desarrollarse a medida que se debilitan los músculos, pero la terapia física regular diseñada para mantener un rango de movimientos en las articulaciones puede ayudar a combatir este problema.
Escoliosis: Los músculos débiles del torso pueden llevar a la curvatura de la columna, o escoliosis, la cual a su vez puede limitar la movilidad e interferir con la respiración. La terapia física regular puede ayudar a retrasar o prevenir la escoliosis, pero con el tiempo puede necesitarse cirugía correctiva.
Debilidad muscular: Los aparatos ortopédicos para las piernas o una silla de ruedas pueden ser necesarios a la larga para ayudar con la movilidad. Un terapeuta ocupacional puede ayudar a las personas con CMD a encontrar la mejor forma de realizar sus funciones cotidianas, a menudo a través del uso de dispositivos de asistencia.
Insuficiencia respiratoria: Los síntomas de la insuficiencia respiratoria incluyen jaquecas matutinas, fatiga, falta de sueño, voz debilitada o suavizada y tos improductiva. La debilidad avanzada o severa en los músculos respiratorios (el diafragma y los músculos de las costillas) puede interferir con la respiración. Hay muchas opciones disponibles para ayudar con este problema, desde ventilación nocturna no invasora hasta una traqueotomía.
Discapacidades de aprendizaje: Algunos niños con CMD pueden tener discapacidades de aprendizaje significativas o retraso mental. Los programas de educación especial, iniciados lo antes posible, pueden ayudar al niño a maximizar su potencial de aprendizaje.
Convulsiones y problemas de la vista: Los especialistas pueden tratar estos problemas con una variedad de terapias.
tapa
DISTROFIA MUSCULAR DISTAL
¿Qué Causa la DD? | ¿Cuáles Son los Tipos de DD? | Problemas y Soluciones en la DD

Descrita por primera vez en 1902, la distrofia muscular distal (DD), o miopatía distal, es el nombre de un grupo de trastornos que afectan principalmente los músculos distales, aquellos que están más lejos de las caderas y los hombros, tales como los músculos de las manos, los pies, los antebrazos y la parte inferior de las piernas. Aunque la debilidad muscular generalmente se detecta primero en los músculos distales, con el tiempo se verán afectados también otros grupos de músculos. El intelecto no es afectado en estas enfermedades.
¿Qué Causa la DD?
Las DDs son causadas por muchos defectos genéticos diferentes, de los cuales todavía no se conocen todos. Además, algunas de las DDs han recibido diferentes nombres basados en varios síntomas, pero pueden en realidad ser causadas por defectos en el mismo gene.
Su propia forma de DD puede o no pertenecer a una de estas categorías. Muchas de estas enfermedades pueden variar de una persona a otra y, en algunos casos, los investigadores están todavía en el proceso de descubrir qué síntomas están vinculados con un defecto genético particular.
¿Cuáles Son los Tipos de DD?
Miopatía distal de Welander
Esta forma de distrofia muscular sigue un patrón hereditario dominante y generalmente tiene su inicio entre los 40 y 50 años de edad. Las extremidades superiores tienden a ser afectadas primero y después las inferiores. El grado de debilidad muscular alcanzado puede variar desde benigno hasta severo. Aunque todavía no se conoce su causa, el trastorno puede estar vinculado con el gene disferlina, el mismo gene que se encuentra defectuoso en la miopatía de Miyoshi y la DM 2B del anillo óseo.
| Clasificación de Distrofias Musculares Distales (DDs) |
| Enfermedad |
Cromosoma/Gene |
Patrón Hereditario |
| Miopatía distal de Welander |
cromosoma 2 |
dominante |
| Miopatía distal finlandesa/Markesbery |
cromosoma 2 |
dominante |
| Miopatía distal de Miyoshi |
disferlina |
recesivo o esporádico |
| Miopatía distal de Nonaka |
cromosoma 9 |
recesivo o esporádico |
| Miopatía distal de Gower |
cromosoma 14 |
dominante |
| Miositis hereditaria de cuerpo de inclusión (HIBM) |
gene GNE |
recesivo |
| Miositis hereditaria de cuerpo de inclusión (HIBM) |
desconocido |
dominante |
| Miopatía distal con debilidad en las cuerdas vocales y la faringe |
cromosoma 5 |
dominante |
Miopatía distal finlandesa/Markesbery
La distrofia muscular de Markesbery sigue un patrón hereditario dominante, con la debilidad comenzando después de los 40 años de edad en las extremidades inferiores y progresando lentamente a los músculos de las extremidades superiores y del torso. Los problemas cardíacos pueden ser característicos.

Los aparatos ortopédicos para las piernas pueden sostener los músculos atrofiados (degenerados) de la parte inferior de las piernas, causados por la DM distal. |
La distrofia muscular finlandesa (también llamada DM de la tibia) puede ser severa o benigna y típicamente afecta solamente a personas de ascendencia finlandesa. Aquellos con solamente un gene defectuoso experimentan debilidad leve en los músculos tibiales de la pierna (parte delantera de la pantorrilla) en algún momento después de los 40 años de edad. Aquellos con dos genes defectuosos tienen debilidad progresiva comenzando en la niñez y pueden perder la habilidad de caminar alrededor de los 30 años de edad.
La miopatía distal finlandesa y la de Markesbery pueden ser causadas por defectos en el mismo gene.
Miopatía distal de Miyoshi
Este trastorno se hereda en un patrón recesivo e involucra debilidad que comienza en las extremidades inferiores, especialmente en el músculo de la pantorrilla. También puede progresar a otros músculos. Los síntomas generalmente comienzan entre los 15 y los 30 años de edad.
El defecto genético que causa la miopatía de Miyoshi ha sido relacionado con el gene de una proteína con función desconocida llamada disferlina. Los defectos en el gene de la disferlina también pueden causar la distrofia muscular 2B del anillo óseo, la cual produce debilidad muscular en el área de las caderas y los hombros. Las personas con el mismo defecto genético en sus genes de disferlina pueden tener una u otra enfermedad y no se sabe qué determina el patrón de síntomas que tendrá una persona.
Miopatía distal de Nonaka
Encontrada usualmente en familias de ascendencia japonesa, esta forma de distrofia muscular distal se hereda en un patrón recesivo y los síntomas comienzan entre los 20 y los 40 años de edad. Los músculos anteriores de la parte inferior de la pierna (aquellos en el frente de ésta) típicamente se ven afectados primero, pero la enfermedad puede progresar hasta afectar los músculos de la parte superior de los brazos y las piernas, así como los músculos del cuello. Los músculos cuadríceps (en el muslo) tienden a mantenerse fuertes.
La enfermedad puede ser causada por un defecto en el mismo gene que causa la miositis hereditaria recesiva de cuerpo de inclusión.
Miopatía distal de Gower
Este trastorno se hereda en una forma dominante y tiene su inicio desde la niñez hasta los 25 años de edad. La debilidad se presenta primero en los músculos de la pierna y del cuello y progresa lentamente hasta incluir los músculos de la parte superior de las piernas, los de las manos y músculos adicionales del cuello.
Miositis hereditaria de cuerpo de inclusión (HIBM)
Este trastorno puede heredarse como una enfermedad dominante o recesiva. La forma recesiva aparece en la adolescencia o entre los 20 y los 30 años de edad y la debilidad muscular se presenta tanto en los músculos distales (aquellos más alejados de los hombros y las caderas) como en los músculos proximales (los músculos de los hombros y de las caderas).
La forma dominante tiene su inicio entre los 25 y los 40 años de edad y la debilidad ocurre en los músculos distales y proximales de las extremidades, con progresión lenta. En ambas formas de HIBM el tejido muscular, cuando se ve en un corte seccional delgado, se caracteriza por la presencia de agujeros minúsculos en las fibras musculares, llamados vacuolas.
Miopatía distal con debilidad en las cuerdas vocales y la faringe

Un terapeuta ocupacional puede enseñarle a las personas con DD cómo desarrollar actividades a pesar de los músculos débiles de los brazos. |
Este trastorno se hereda en un patrón dominante y ha sido vinculado con el cromosoma 5, en la misma región que el gene que se encuentra defectuoso en la DM tipo 1A del anillo óseo. Los síntomas aparecen por primera vez entre los 35 y los 60 años de edad e incluyen debilidad en las manos, las piernas o la voz. La dificultad al tragar, disfagia, puede ser una característica.
Problemas y Soluciones en la DD
•Debilidad en las extremidades inferiores: La debilidad en los músculos de las extremidades inferiores puede hacer difícil caminar o el levantarse después de estar sentado. En algunos casos, un tipo de aparato ortopédico llamado ortosis, que se usa encima del zapato y de la parte inferior o superior de la pierna, puede ayudar con la debilidad en las piernas. Con el tiempo, podría necesitarse una silla de ruedas.
•Debilidad en los brazos: Su clínica MDA puede enviarlo con un terapeuta que le ayudará a sacar el mayor provecho a sus músculos de los brazos para desarrollar sus actividades cotidianas. A menudo, el te-rapeuta puede recomendar dispositivos que pueden mejorar la fuerza prensil, o ayudarle a levantar sus brazos para desarrollar mejor algunas actividades como lavarse los dientes o peinarse.
tapa
DISTROFIA MUSCULAR DE EMERY-DREIFUSS (EDMD)
¿Cómo se Hereda la EDMD? | ¿Qué Causa la EDMD?
¿Qué le Sucede a Alguien con EDMD? | Problemas y Soluciones en la EDMD

La distrofia muscular de Emery-Dreifuss se caracteriza por la degeneración y debilidad en los músculos que forman los hombros y la parte superior de los brazos, y los músculos de la pantorrilla en las piernas. Otro aspecto predominante de esta enfermedad es la presencia, muy temprano en el curso de la enfermedad, de contracturas (articulaciones rígidas) en los codos, la nuca y los talones. Por último, y muy importante, es común encontrar en la EDMD un tipo de problema del corazón llamado bloqueo de conducción, el cual necesita ser monitoreado.
¿Cómo se Hereda la EDMD?
La forma más común de EDMD se hereda en un patrón X-ligado, pero la EDMD también puede heredarse de una manera dominante y, muy raramente, de una manera recesiva. Aunque las formas de EDMD X-ligada, dominante y recesiva siguen patrones hereditarios diferentes, sus síntomas son casi imposibles de distinguir.
¿Qué Causa la EDMD?
Recientemente, los investigadores han identificado los genes que, cuando son defectuosos, conducen a las diferentes formas de EDMD. Ahora sabemos que el gene que se presenta defectuoso en la EDMD X-ligada produce una pequeña proteína llamada emerina, la cual normalmente se encuentra en la membrana que rodea el núcleo de cada célula (el compartimiento que contiene los cromosomas en el centro de una célula).
Todavía no se sabe cómo es que la pérdida de emerina en la membrana de la célula en la EDMD X-ligada conduce a los síntomas de distrofia muscular. Algunos investigadores piensan que la falta de emerina interfiere con la reorganización de la membrana nuclear después que una célula se ha dividido, llevando a células débiles o moribundas.

La EDMD lleva a dificultad para doblar los codos, además de debilidad muscular. |
De forma parecida, el gene que se ha encontrado defectuoso tanto en las formas autosomal como recesiva de EDMD, contiene las instrucciones para dos proteínas íntimamente relacionadas, llamadas lamina A y lamina C, las cuales también están asociadas con la membrana nuclear de las células.
De nuevo, no se sabe aún cómo la pérdida de las laminas A y C lleva a los síntomas de distrofia muscular, pero algunas investigaciones sugieren que la membrana nuclear puede desestabilizarse sin las proteínas laminas, resultando en la descomposición de los músculos.
Otra pregunta que sigue sin respuesta es por qué los síntomas de EDMD se limitan principalmente a los músculos esqueléticos (voluntarios) y los músculos del corazón, siendo que las proteínas emerina y lamina se encuentran en la mayoría de los tejidos del cuerpo.
¿Qué le Sucede a Alguien con EDMD?
Generalmente, los síntomas de la EDMD son visibles a los 10 años de edad, pero el trastorno tiende a progresar lentamente. Las primeras señales incluyen "caminar de puntillas" debido a la rigidez en los tendones de Aquiles, y dificultad en doblar los codos. Más adelante, las señales de debilidad muscular son más prominentes, pero todavía en general se les considera leves. Usualmente, los problemas cardíacos se pueden detectar hacia los 20 años de edad, pero también pueden ocurrir en etapas más tempranas de la enfermedad. El intelecto no se ve afectado.
Las contracturas que ocurren tempranamente en la EDMD pueden dificultar los movimientos de los brazos, el cuello, los talones y la columna vertebral; sin embargo, el progreso de la debilidad muscular parece ocurrir muy lentamente en la EDMD y posiblemente no comience a causar dificultades hasta más tarde en la vida.
Los problemas cardíacos pueden poner en peligro la vida y pueden requerir la inserción de un marcapasos. Algunas mujeres que son portadoras genéticas de la EDMD X-ligada pueden también correr el riesgo de problemas cardíacos y este riesgo puede aumentar con la edad (los portadores de EDMD X-ligada no tienen tendencia a tener debilidad muscular o contracturas).
Problemas y Soluciones en la EDMD
Es importante que las personas con EDMD monitoreen sus funciones cardíacas. |
Contracturas: Las contracturas se desarrollan temprano en la EDMD y pueden empeorar aun cuando no cambie la fuerza muscular. La prevención de las contracturas es difícil, pero mantener el rango de los movimientos mediante terapia física puede ayudar a retardar su desarrollo. El alivio quirúrgico de las contracturas es complicado debido a su tendencia de volverse a desarrollar.
Bloqueo de conducción cardíaca: Esta forma de problema del corazón ocurre cuando el ritmo de las palpitaciones se desestabiliza porque los impulsos eléctricos no se comunican adecuadamente entre la cámara superior y la cámara inferior del corazón. El bloqueo de conducción puede llevar a un paro cardíaco repentino. Para los 30 años de edad, casi todas las personas con EDMD tendrán alguna clase de complicación cardíaca que puede ser detectada.
Afortunadamente, este problema es fácil de detectar mediante un electrocardiograma y la inserción de un marcapasos puede salvar la vida.
En algunos casos, después de la inserción del marcapasos, puede desarrollarse, con el tiempo, una falla cardíaca crónica generalizada. Hay muchos medicamentos disponibles para ayudar a combatir las fallas cardíacas crónicas. Toda persona que reciba un diagnóstico de EDMD debe ser monitoreada regularmente para detectar señales de bloqueo de conducción cardíaca.
tapa
DISTROFIA MUSCULAR OCULOFARINGEA
¿Qué Causa la OPMD? | ¿Qué le Sucede a Alguien con OPMD? | Problemas y Soluciones en la OPMD
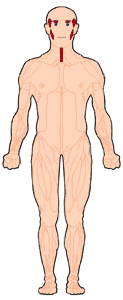
La distrofia muscular oculofaríngea (OPMD) se caracteriza por debilidad en los músculos que controlan los párpados (llevando a párpados caídos, una condición también conocida como ptosis), y por debilidad en los músculos faciales y los músculos de la faringe (aquellos en la garganta utilizados para tragar). También afecta los músculos de las extremidades. Los síntomas de la enfermedad usualmente no comienzan hasta los 40 o 50 años de edad, pero pueden ocurrir antes.
La OPMD generalmente se hereda como una enfermedad dominante, pero casos excepcionales pueden demostrar un patrón hereditario recesivo. Cuando se examinan bajo el microscopio los tejidos musculares de una persona con OPMD, pueden verse aglomeraciones de proteínas llamadas inclusiones en los núcleos celulares de los músculos (los compartimientos celulares que contienen los cromosomas).
La enfermedad es más común en familias francocanadienses o familias de ascendencia francocanadiense. La investigación genealógica de estas familias sugiere que los integrantes de una sola pareja, Zacharie Cloutier y Saincte Dupont, quienes emigraron de Francia al Canadá en 1634, pueden haber albergado el defecto genético responsable de la mayoría de los casos francocanadienses de hoy día. También hay una alta incidencia de OPMD entre los residentes hispanos del norte de Nuevo México.
La OPMD también puede afectar a personas que no son de origen francocanadiense o hispano.
¿Qué Causa la OPMD?

Los párpados caídos son una característica común de la OPMD. |
El gene que se encuentra defectuoso en la OPMD fue descubierto en 1998 y se llama el gene de proteína de enlace poly (A) 2, o PABP2. Los investigadores sospechan que en la OPMD, la presencia de aminoácidos adicionales en la proteína producida por un gene de PABP2 defectuoso, hace que la proteína PABP2 se aglomere en los núcleos de las células musculares, quizá interfiriendo con las funciones celulares. La enfermedad puede diagnosticarse a menudo con una prueba de ADN sobre la mutación más común de PABP2.
No se sabe por qué los síntomas de OPMD generalmente ocurren tarde en la vida o por qué están restringidos a los grupos musculares específicos relacionados con la OPMD.
¿Qué le Sucede a Alguien con OPMD?
Una persona con OPMD puede notar primero sus párpados caídos, que gradualmente llevan a inclinar la cabeza hacia atrás para poder ver bien. Alternativamente, algunas personas pueden notar primero que tienen una tendencia a atragantarse con frecuencia y pueden tener otros problemas relacionados con dificultad para tragar (llamada disfagia). Con el tiempo, la mayoría de las personas desarrollan en algún grado ambas condiciones, tanto ptosis como disfagia.
Es común la debilidad final de los músculos de la cara y las extremidades. Por ejemplo, muchas personas con OPMD reportan problemas al arrodillarse, doblarse, acuclillarse, caminar y subir escaleras. También pueden presentarse la visión doble y una condición "velada" en la voz.
Actualmente no hay cura para la OPMD, pero muchas personas con la enfermedad encuentran beneficioso el tratamiento de los síntomas conforme ocurren.
Problemas y Soluciones en la OPMD
Disfagia: La dificultad para tragar, o disfagia, puede hacer que una persona aspire comida o líquido hacia los pulmones, lo cual a su vez puede llevar a un problema serio llamado neumonía por aspiración. Si usted encuentra que se atraganta frecuentemente mientras come o bebe, puede ser necesario que un profesional evalúe sus habilidades para tragar.
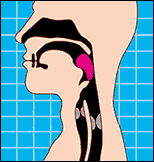
La OPMD puede causar debilidad en los músculos utilizados para tragar, lo que puede llevar a atragantarse con frecuencia. |
Hay una cantidad de técnicas que pueden ayudar a tratar la disfagia, desde mantener la cabeza en diferentes posiciones hasta el uso de aditivos comerciales para espesar los líquidos y darles una consistencia manejable. En casos avanzados, puede justificarse un procedimiento no quirúrgico llamado estiramiento de la garganta o un procedimiento quirúrgico llamado miotomía cricofaríngea. La alimentación por medio de sondas es otra opción para casos avanzados.
Conforme sea necesario, su clínica MDA lo enviará con un patólogo de lenguaje-dicción (SLP) o un otorrinolaringólogo (doctor de oído, nariz y garganta).
Ptosis: Los párpados caídos, o ptosis, pueden reducir significativamente la visión y conducir a incomodidad social. Este problema se puede resolver mediante un tipo de cirugía de reducción de los párpados llamado soporte frontal, llevado a cabo por un cirujano oculoplástico.
Debilidad en las extremidades: La dificultad para levantar los pies mientras se camina puede llevar a tropiezos y caídas. Pueden ser útiles los aparatos ortopédicos para las piernas, los bastones o los andadores. A la larga, las personas con OPMD pueden requerir una silla de ruedas para hacer más conveniente su movilidad.
La debilidad en la parte superior de los brazos y en los hombros que limita las funciones puede tratarse con técnicas de adaptación a través de la terapia ocupacional.
tapa
¿QUE EXAMENES SE UTILIZAN PARA DIAGNOSTICAR LA DISTROFIA MUSCULAR?
En el diagnóstico de cualquier forma de distrofia muscular, un doctor generalmente comienza recabando la historia del paciente y su familia y haciendo un examen físico. Se puede obtener mucha información así, incluyendo el patrón de debilidad. La historia y el examen físico permiten avanzar mucho hacia el diagnóstico, aun antes de hacer cualquier prueba complicada de diagnóstico.
El médico también desea determinar si la debilidad del paciente se debe a problemas en los músculos como tales o en los nervios que los controlan. Los problemas con los nervios que controlan los músculos, o nervios motores, que se originan en la médula espinal y se extienden a todos los músculos, pueden causar debilidad que se parece a un problema muscular pero en realidad no lo es.
Usualmente, el origen de la debilidad puede ser determinado mediante un examen físico. Ocasionalmente, se hace una prueba especial llamada electromiografía (EMG). En esta clase de prueba, se utilizan la electricidad y agujas muy finas para estimular individualmente los músculos o nervios para ver dónde se encuentra el problema. La electromiografía es incómoda, pero generalmente no es muy dolorosa.
Durante el inicio del proceso de diagnóstico, los doctores a menudo ordenan un examen de sangre especial llamado nivel de CK. Las letras CK identifican a la creatina quinasa, una enzima que se fuga de los músculos dañados. Cuando se encuentran niveles elevados de CK en una muestra de sangre, generalmente significa que los músculos están siendo destruidos por algún proceso anormal, como una distrofia muscular o una inflamación. Un nivel alto de CK, por lo tanto, sugiere que los músculos en sí son la causa probable de la debilidad, pero no indica exactamente cuál pueda ser el trastorno muscular.
Para determinar cuál trastorno está causando el aumento de CK, el médico puede ordenar una biopsia muscular, la remoción quirúrgica de una pequeña muestra de músculo del paciente. Al examinar esta muestra, los doctores pueden obtener mucha información acerca de lo que actualmente sucede dentro de los músculos. Las técnicas modernas pueden utilizar la biopsia para distinguir las distrofias musculares y separarlas de infecciones, trastornos inflamatorios y otros problemas.
Otros exámenes de la muestra de biopsia pueden proporcionar información acerca de cuáles proteínas de los músculos se encuentran en las células musculares, y si están presentes en cantidades normales y en los lugares correctos. Esto puede decirle al médico y al paciente qué anda mal con las proteínas de las células y dar a conocer candidatos probables en cuanto a los genes responsables del problema. Sin embargo, la correlación entre las proteínas faltantes en la biopsia muscular y los defectos genéticos no es perfecta. Un médico de la clínica MDA puede ayudarle a entender estos resultados.
También puede ordenarse un rastreo de resonancia magnética (MR). Estos procedimientos, que son sin dolor, permiten a los doctores visualizar qué está ocurriendo dentro de los músculos que se están debilitando.
Las pruebas genéticas, utilizando una muestra de sangre, pueden analizar los genes de una persona en busca de defectos particulares que causan las distrofias musculares poco comunes, pero estas pruebas a menudo no son necesarias para el diagnóstico o para determinar el tratamiento.
tapa
LA BUSQUEDA DE TRATAMIENTOS Y CURAS DE LA MDA
Desde que las investigaciones pioneras de 1986 permitieron a Louis Kunkel, científico apoyado por la MDA, identificar al gene involucrado en la distrofia muscular de Duchenne, los investigadores de la MDA han avanzado en el aislamiento y caracterización de los genes involucrados en casi todos los trastornos neuromusculares cubiertos por el programa de la MDA. Estos incluyen muchos de los defectos genéticos responsables de los trastornos descritos en este folleto. Estos descubrimientos le han permitido a los científicos entender las variaciones entre las diferentes formas de las enfermedades y han ayudado a los médicos a proporcionar diagnósticos más acertados.
| El sitio Web de la MDA is constantemente actualizado con la información más reciente sobre las enfermedades neuromusculares en su programa. Lea las más recientes noticias de investigación en inglés o español. |
Ahora que este primer paso esencial está casi terminado en su totalidad, la MDA está explorando formas de corregir los problemas musculares causados por los diferentes defectos genéticos. Las áreas de investigación activa incluyen:
Terapia genética: Un mecanismo para suplir los genes defectuosos con genes sanos en los tejidos afectados por enfermedades neuromusculares
Reparación genética: Un mecanismo para reparar el defecto genético en el propio ADN de una persona en las células de los tejidos afectados
Terapia celular: Un mecanismo para trasplantar tejido muscular sano de repuesto, utilizando células primitivas llamadas células madre de un donante sano. En el futuro, la terapia celular podrá ser utilizada en combinación con la terapia genética para corregir defectos en las propias células madre musculares de una persona.
En septiembre de 1999, la MDA lanzó sus primeras pruebas clínicas humanas de terapia genética para probar qué tan seguro es reemplazar genes defectuosos en cuatro formas específicas de distrofia muscular del anillo óseo. Actualmente se encuentran en etapa de planificación las pruebas de terapia genética y terapia celular para otros trastornos.
LA MDA ESTA AQUI PARA AYUDARLE
La Asociación de la Distrofia Muscular ofrece una amplia gama de servicios para ayudarle a usted y a su familia a hacerle frente a todas las formas de distrofia muscular. Ya sea que usted sea un adulto que acaba de recibir el diagnóstico de una distrofia muscular poco común, o el padre o madre de un hijo con tal enfermedad, el personal de su oficina local de la MDA está allí para ayudarle en muchas formas. Los servicios de la Asociación incluyen lo siguiente:
- una red nacional de 220 clínicas afiliadas a hospitales con los mejores especialistas en enfermedades neuromusculares
- campamentos de verano de la MDA, de una semana de duración, para niños con enfermedades neuromusculares
- grupos de apoyo para las personas afectadas, sus cónyuges, padres de familia u otras personas que las cuiden
- ayuda con la compra y reparación de sillas de ruedas y ortosis para las piernas
- evaluaciones de terapias física, ocupacional y respiratoria
- vacunas contra la gripa para proteger el sistema respiratorio
- préstamo de equipo
La MDA le ayuda a mantenerse al tanto respecto a novedades en la investigación, hallazgos médicos e información de discapacidad a través de oradores educativos, seminarios, videos, boletines informativos y más. Asegúrese de pedirle a su oficina local algunos de los folletos más recientes de la MDA, incluso "Servicios para la Persona, la Familia y la Comunidad". También querrá obtener ejemplares de "Respire Mejor: Cuidado respiratorio para niños con distrofia muscular", "Aprendiendo a Vivir con una Enfermedad Neuromuscular, Un Mensaje para los Padres", y algunas publicaciones para niños. Muchos folletos de la MDA están disponibles en español. Todos los que están inscritos en la MDA reciben también Quest, la revista nacional de la MDA.
La página electrónica mundial de la MDA, en www.mda.org, contiene más de 1.200 páginas de información valiosa, incluyendo los textos de artículos de números pasados de Quest y otras publicaciones de la MDA.
Si tuviera alguna pregunta acerca de las distrofias musculares, alguien en la MDA le ayudará a encontrar la respuesta.
tapa
|