|
|
| Studies |
 |
|
|
Rembrandt Application
REMBRANDT Home 
Welcome to REMBRANDT (Repository of Molecular
Brain Neoplasia Data)
AboutMotivation and background
Glioma Molecular Diagnostic Initiative’s (GMDI) primary goal is to develop a molecular
classification schema that is both clinically and biologically meaningful, based on gene
expression and genomic data from tumors (Gliomas) of patients who will be prospectively
followed through natural history and treatment phase of their illness. Secondary objective
of this study is to explore gene expression profiles to determine the responsiveness of
the patients and correlate with discrete chromosomal abnormalities. 
REMBRANDT Knowledgebase ProtocolsProcessSamples are collected from patients enrolled in the GMDI study. The following illustrations indicate what samples are being collected and how they are processed.
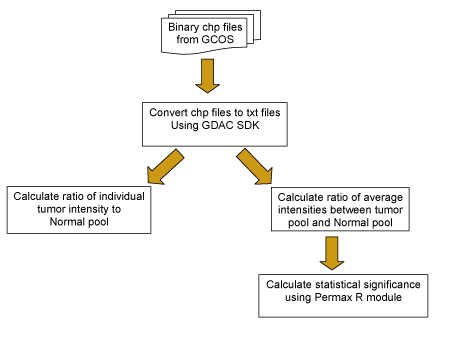
Data ProcessingGene expression data was collected using both Affymetrix and cDNA array platforms. For the Affymetrix experiments, Human Genome U133 Plus 2.0 Arrays were used to hybridize tumor samples collected from patients with brain tumor. The data from the hybridizations were used to compare the levels of expression between normal and brain tumor samples.>/p> For data preprocessing, the probe level data was consolidated into probseset data using Affy MAS5 algorithm, with target scaling value at 500. We also processed probe level data with custom CDF (Chip Definition Files) that rearrange Affymetrix probes into gene-based probe sets. Probes mapped to alternatively spliced exons were grouped into a distinct probesets. The most 3` probes were selected for processing. Non-specific probes were masked before processing. Single tumor sample to normal pool and sample average (samples were averaged based on tumor subtypes into 6 categories, Glioblastoma Multiforme, Oligodendroglioma, Astrocytoma, Mixed, Unclassified and Unknown tumors) to normal pool comparison were performed. The group comparisons were performed in R with two sample t tests. The signal values were first transformed to logarithm (base 2). The averages of the log2-signals of tumor and normal groups were computed. The magnitude of the differences between the geometric means of expression levels for each reporter from the two groups was computed. The significance of the differences between tumors (or each subtype of tumor) and the normal samples for each reporter was also evaluated. For each individual tumor sample, signals for each tumor and ratio between each tumor and average of normal (geometric means, computed the same way as described above) were computed. Affymetrix data analysis workflow is briefly shown below. All the processes were performed separately for various data groups (public data and institution-based data).
Home-grown “Glioma Microarray chip” developed in conjunction with collaborators in the Cancer Genome Anatomy Project (CGAP) containing approximately 50,000 IMAGE clones that are of relevance to tumor development, was used. The arrays were scanned and processed using GenePix image analysis software and had been previously normalized (Lowess normalization) and filtered, thus contains missing values.
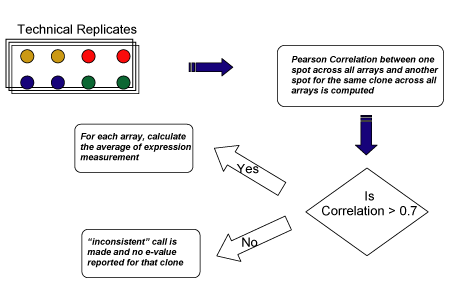
For clones with multiple spots (well ids) on the chip, a correlation-based approach was used to obtain a consolidate expression value for each clone. First, Pearson correlation between expression measurements (log2-ratios) for one spot (with a unique well id) for all arrays and those for another spot with the same clone was computed. If the correlation was above a threshold (e.g. 0.7), then, for each array, the average of the expression measurements among these spots was computed to represent the expression value for that clone. If the correlation is under a threshold (e.g. 0.7), the expression measurements between the spots from the same clone were considered to be inconsistent, and an "inconsistent" call was made and no final expression values were provided for that clone. If there were more than two spots from the same clone on the chip, pair-wise correlations between expression values (log2-ratios) of different spots (with different well ids) from the same clone were computed. Currently, if any of the correlation was under the threshold, an "inconsistent" call was made; otherwise the average of the expression measurements was computed to represent expression for that clone for each array. cDNA array data processing workflow to handle replicates is briefly shown below.
For single array/tumor values, if a value (log2-ratio) was missing in the input data, then ratio between the tumor and normal for that clone will be missing. For the group comparison, for each clone, the available (non-missing) data was used to compute the average and p-value. If for a particular tumor subtype, the data was missing for all of the arrays in that subtype for a particular clone, then the average and p-value will both be missing in the final result.
LPG/Unified Gene Algorithm was developed by NCI’s Laboratory of Population Genetics. The algorithm provides a gene-based view of the expression data. To obtain the unified gene expression values, the probe-level data is processed with custom CDF (Chip Definition Files) that rearranges Affymetrix probes into splice-form based probesets. Probes mapped to alternatively spliced exons are grouped into a distinct probeset. The most 3` probes are selected for processing. Non-specific probes are masked before processing. To obtain Copy number information, the tumor samples were hybridized to Affymetrix 100K SNP arrays. The CHP files from the Affymetrix Gene Chip Operating System were processed using the GDAS3.0 (GeneChip® DNA Analysis Software)and CNAT (Copy number Analysis Tool2.1). The copy number data was collected for each mapping SNP reporter on the Chip, for all the tumor samples. The clinical data was collected by electronic data management system hosted by MD Anderson. The reports were created by querying that database and exported as flat files. The files were loaded into the stage area of the caIntegrator data warehouse. After going through some data quality checks as well as required transformation, the data was loaded into the data warehouse tables and available via the Rembrandt data portal. News
in collaboration with:

|
|
|