ISSN: 1080-6059
Volume 14, Number 3–March 2008
Research
High Rate of Mobilization for blaCTX-Ms
Miriam Barlow,* ![]() Rebecca A. Reik,† Stephen D. Jacobs,* Mónica
Medina,* Matthew P. Meyer,* John E. McGowan, Jr.,† and Fred C. Tenover‡
Rebecca A. Reik,† Stephen D. Jacobs,* Mónica
Medina,* Matthew P. Meyer,* John E. McGowan, Jr.,† and Fred C. Tenover‡
*University of California, Merced, California, USA; †Emory
University, Atlanta, Georgia, USA; and ‡Centers for Disease Control and
Prevention, Atlanta, Georgia, USA
Suggested citation for this article
Abstract
We constructed a phylogenetic analysis of class A β-lactamases
and found that the blaCTX-Ms have been mobilized to plasmids ≈10 times more frequently than other class A β-lactamases.
We also found that the blaCTX-Ms are descended from a common
ancestor that was incorporated in ancient times into
the chromosome of the ancestor of Kluyvera species through horizontal
transfer. Considerable sequence divergence has occurred among the descendents
of that ancestral gene sequence since that gene was inserted. That divergence
has mainly occurred in the presence of purifying selection, which indicates a
slow rate of evolution for blaCTX-Ms in the pre–antimicrobial
drug era.
Antimicrobial drug–sensitive bacteria become resistant to antimicrobial drugs through a variety of mechanisms, such as chromosomal mutations that up-regulate the expression of antibiotic-resistance genes, DNA uptake through transformation, or the process of conjugation. The ability of plasmids to evolve independently of their hosts has allowed numerous resistance genes from diverse species of bacteria to assemble within single plasmids and spread into a wide variety of organisms (1). The mobilization of a chromosomal resistance gene to a plasmid is an important event because the mobilized gene is now capable of spreading widely throughout diverse species of bacteria and because the fitness advantage that a plasmid confers generally increases as it acquires more resistance genes (1).
The class A β-lactamases have been the most frequently encountered plasmidic resistance genes. Class A β-lactamases from the TEM group have occurred at a particularly high frequency; in many surveillance studies, they have been identified as the resistance determinants most frequently encountered (2–9). The first blaTEM allele, blaTEM-1, is a plasmidic allele that was first isolated in 1963 (10,11). Currently, ≈160 different plasmidic alleles encode unique TEM β-lactamase enzymes (www.lahey.org/Studies), and all are descended from a single plasmidic ancestor, blaTEM-1 (12).
The SHVs are another group of class A β-lactamases that have been frequently observed in surveillance studies. As with the TEMs, numerous alleles encode unique SHV enzymes (≈105), and the SHVs are all descended from a single ancestor (13). The first blaSHV allele was detected in 1974 (10,14). Unlike blaTEMs, the blaSHVs are present in the chromosome of nearly all Klebsiella pneumoniae isolates belonging to the KP1 group. Evidence suggests that blaSHVs have been chromosomally located since the pre–antimicrobial drug era (15), and they may have been mobilized to plasmids up to 4 times, although the sequence divergence among them is insufficient to clearly resolve the independent mobilizations of the blaSHVs.
The CTX-Ms are another group of class A β-lactamases that are located on plasmids and that have been of particular clinical interest because they are rapidly spreading through clinical populations of bacteria. The first plasmidic blaCTX-M observed in human-associated clinical populations was isolated in 1989 (16,17). Unlike the usual pattern of class A β-lactamase mobilizations in which the plasmidic alleles are all descended from a single common plasmidic ancestor, evidence shows that CTX-Ms have been mobilized numerous times from the chromosomes of Kluyvera (16,18–21). Because Kluyvera chromosomal genes have been found that exactly match the sequence of plasmidic CTX-Ms (18), many of the mobilizations have likely occurred recently. To investigate the mobilization of the blaCTX-Ms to plasmids, we generated a phylogenetic analysis of the CTX-Ms that included a representative sampling of other class A β-lactamases.
Methods
BLAST Search
blaCTX-M and blaCTX-M homologue DNA sequences were identified with a TBLASTN (www.ncbi.nlm.nih.gov/blast) (22,23) search of the nonredundant National Center for Biotechnology Information (NCBI) sequence database and the completed microbial genomes database by using characterized blaCTX-Ms as query sequences (blaCTX-M1 and blaCTX-M2). The BLAST search of the completed microbial genomes identified positive matches for organisms that contain blaCTX-M homologs. The BLAST search of completed genomes also showed which microbes have no close blaCTX-M homologue, and thus enabled horizontal transfer events to be identified.
Alignment
The protein sequences of the blaCTX-Ms and their homologs were aligned with ClustalX 1.8 (24) by using the Gonet 250 similarity matrix with a gap-opening penalty of 35 and a gap-extension penalty of 0.75 for the pairwise alignment stage, and a gap-opening penalty of 15 and a gap extension penalty of 0.3 for the multiple alignment stage. The corresponding DNA coding sequences were aligned by introducing triplet gaps between codons corresponding to gaps in the aligned protein sequences with the CodonAlign program. (CodonAlign for Macintosh and for PC [Windows] computers and source code that can be compiled for other platforms are available at no charge from http://sinauer.com/hall.)
Estimation of Positive Selection
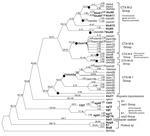 |
Figure 1. Phylogenetic analysis of blaCTX-Ms. This tree was calculated by Bayesian inference. Number of mutations occurring along each branch are given along the length of the branch... |
 |
Figure 2. Phylogenetic analysis of class A β-lactamases calculated by Bayesian inference. Number of mutations occurring along each branch are represented visually by the lengths of the branches... |
Estimation of the nonsynonymous (dN) and synonymous (dS) substitution rates is an important means of understanding mechanisms of molecular evolution. A dN/dS ratio >1 is taken as evidence of positive selection, whereas a dN/dS ratio <1 is taken as evidence of purifying selection (25). The Codeml program of the PAML package (available from http://envgen.nox.ac.uk/bioinformatics/docs/codeml.html) (25) was used to estimate dN/dS ratios in the phylogenetic analysis shown in Figure 1. The values were calculated by using model 1 in the program, and default parameters were used for the execution of the program.
Phylogenetic Reconstruction
Phylogenies were constructed by the Bayesian method, as implemented by the program MrBayes (26) (available at no charge from www.mrbayes.net). The evolutionary model used was the General Time Reversible model (27). Because evolutionary rates are not homogeneous for every site in a gene, among-site variation in evolutionary rate was estimated separately for first, second, and third positions of sites within codons. Four chains, with a "temperature" of 0.2 for the heated chains, were run for each tree. Trees were sampled every 100 generations. A total of 10 million generations were run with a burn-in of 5,000 trees. The length of each burn-in was set at a value that exceeded twice the number of trees required for convergence upon a stable likelihood value. Because the consensus trees calculated by MrBayes do not include the posterior probabilities of the clades, each entire set of trees was imported into PAUP* (28) and the same trees used by MrBayes to calculate a consensus were used to calculate a 50% majority rule consensus in PAUP* (28). The resulting tree shows the posterior probabilities of the clades, i.e., the percentage of time that those taxa are included in the clade. The consensus trees calculated by MrBayes were imported into PAUP* for the purposes of displaying and printing the tree.
Results
Ancient Horizontal Transfer of blaCTX-M Ancestor
The NCBI genomes database (www.ncbi.nlm.nih.gov:80/entrez/query.fcgi?db=genome) currently contains the completed genomic sequences of 139 eubacterial organisms. Because Kluyvera are members of the Enterobacteriaceae group of the gamma subdivision of Proteobacteria, the genomes of other members of the gamma subdivision, and especially the chromosomes of Enterobacteriaceae, were of the greatest interest. BLAST searches of these microbial genomes show that among the complete genomic sequences available for 9 species of Enterobacteriaceae (Escherichia coli, Salmonella typhimrium LT2, S. enterica, Shigella flexneri, Photorhabdus luminescens, Buchnera aphidicola, Candidatus Blochmannia floridanus, Wigglesworthia glossinidia, and Yersinia pestis) none contain chromosomal homologs of the blaCTX-Ms that are detectable through sequence comparison. BLAST searches similarly show that many non-Enterobacteriaceae members of the gamma subdivision of Proteobacteria, Shewanella oneidensis, Haemophilus ducreyi, H. influenzae, Pseudomonas aeruginosa, P. putida, P. syringae, Vibrio cholerae, V. parahemolyticus, V. vulnificus, Xanthomonas axonopodis, X. campestris, and Xylella fastidiosa, also do not contain chromosomal homologs of the blaCTX-MS. However, BLAST searches of the translated nonredundant nucleotide database revealed that Kluyvera species, Citrobacter sedlakii, and Klebsiella oxytoca contain close chromosomal homologs of the blaCTX-MS.
These results show that the blaCTX-M homologs originally came into the chromosomes of K. oxytoca, Kluyverra species, and C. sedlakii by horizontal transfer because most species of gamma Proteobacteria do not contain blaCTX-M homologs. If blaCTX-M homologs were vertically transmitted into the species that contain them, numerous deletions would be required to explain absence of those homologs in the majority of gamma Proteobacteria. However, only 3 horizontal insertions are required to explain the presence of blaCTX-M homologs in the chromosomes of K. oxytoca, Citrobacter species. Because fewer insertions than deletions are required to explain these data, insertion of blaCTX-M homologs into the chromosomes of those bacteria that contain them is the most likely explanation for their current distribution.
Divergence of the blaCTX-Ms
The GenBank DNA and protein accession numbers of the sequences included in this analysis are shown in the Appendix Table, along with the organism in which the gene exists and whether the gene was located on a plasmid or a chromosome. The results of our phylogenetic analysis are presented in Figure 1. The groupings of blaCTX-Ms on our phylogenetic tree agree with the dendrogram published by Bonnet in a recent review (16); for purposes of clarity, we will use the same group names used in that review, as shown in Figure 1.
The phylogenetic analysis shows that the blaCTX-Ms represent a fairly divergent group of β-lactamase genes descended from a common ancestor. The genes encoding the CTX-M-1 and CTX-M-2 groups are separated by over 400 mutations, which indicates considerable diversification of the blaCTX-Ms. The average distance separating the blaCTX-Ms from their most recent common ancestor is 226.2-nt ± 22.8-nt mutations, which indicates that the rates of evolution among the blaCTX-Ms have been similar.
Positive selection testing within the phylogenetic analysis shows that positive selection has occurred throughout the evolutionary history of the class A β-lactamases. More positive selection appears to exist at branches deep within the tree than along more recent branches. The branches during which most of the divergence of the blaCTX-Ms occurred are characterized by purifying selection. The detection of purifying selection suggests a slow evolutionary rate and that the blaCTX-Ms diverged in ancient times. More recent evolution of the blaCTX-Ms likely can be characterized by intense positive selection, but the branches at the tips are still too short to obtain reliable dn/ds ratios.
Mobilization of blaCTX-MS to Plasmids
The blaCTX-Ms have been mobilized from the chromosomes of various Kluyvera species to plasmids at least 8 times since they diverged from their most recent common ancestor as indicated in Figure 1. The alleles in the CTX-M-2 group have been mobilized from the chromosome of Kluyvera ascorbata at least twice (29). The alleles in the CTX-M-9 group have been mobilized once from the chromosome of Kluyvera georgiana (30). The alleles from the CTX-M-8 group were mobilized once from the chromosome of K. georgiana (20). The CTX-M-25 group has been mobilized once, although the species from which it originates has not yet been determined. The alleles in the CTX-M-1 group have been mobilized at least 3 times (17,18,31), and one of those mobilizations has been from the chromosome of K. ascorbata. When compared with the blaTEMs, which have been mobilized once, and the blaSHVs, which have been reported to have been mobilized 2–4 times (32), the number of mobilization events that have occurred among the blaCTX-Ms is atypically high.
To compare the number of mobilizations that have occurred in the CTX-M group with those that have occurred in the rest of the class A β-lactamases, we constructed a phylogenetic analysis of class A alleles that spans the breadth of this group and that contains representatives of all groups of class A alleles known to the authors (Figure 2). Among all of the class A β-lactamases, including the blaCTX-Ms, only 22 mobilizations to plasmids were found. To quantitatively compare the numbers of times that CTX-Ms have been mobilized to plasmids with the number of times that other class A β-lactamases have been mobilized to plasmids, the total number of mutations that have occurred within the blaCTX-M clade were summed and divided by the number of mobilizations that have occurred in that region of the phylogenetic analysis. Among the blaCTX-Ms, the ratio of mobilizations to mutations is 1 mobilization per 191 mutations. Among the remainder of the tree when the blaCTX-M clade is excluded from the analysis, 14 mobilizations occur with the ratio of mobilizations to mutations being 1 mobilization per 2,471 mutations. When the complete phylogenetic analysis is considered, 1 mobilization occurs per 1,870 mutations. By that comparison, the mobilization of the blaCTX-M genes to plasmids has occurred at an unusually high rate. This result is unlikely to be an artifact of sampling bias or clinical interest because other class A β-lactamases have been intently studied for a longer period than the blaCTX-Ms. If any bias exists in the data, it would be the undersampling of blaCTX-M mobilizations relative to other class A β-lactamases.
Because nearly one half of the mobilizations that have occurred in the class A phylogenetic analysis have occurred among the blaCTX-MS, it seemed reasonable to conclude that the circumstances associated with the mobilizations of the blaCTX-MS may differ from the circumstances associated with the mobilizations of other class A β-lactamases. To rule out any effect that varying intensities of selection or varying evolutionary rates might have on mobilizations to plasmids, we divided the phylogenetic analysis into several monophyletic groups for further analysis. Within the class A β-lactamase phylogenetic tree (Figure 2) are several monophyletic clades that descended from a single ancestor (node A). Each of the monophyletic clades that descended from node A were considered separately during positive selection testing except for the monophyletic clade that contains the blaCTX-Ms; it was divided into 2 separate clades so that a clade containing only the blaCTX-Ms and their closest relatives could be considered. Monophyletic clades that diverged before the point represented at node A were also examined individually.
The dn/ds ratios were computed for each clade (Table), and a correlation coefficient for mobilizations and dn/ds ratios of 0.21 (p = 0.40) was calculated. The average distance of each clade from the root of the tree was also computed (Table), and the correlation coefficient for mobilizations and average distance from the root is 0.21 (p = 0.41). The nonsignificant p values yielded by those results mean that the unusually high number of mobilizations among the blaCTX-Ms are probably not an artifact caused by positive selection or evolutionary rate.
Most of the mobilizations of the blaCTX-Ms have occurred in recent years because genes that are identical (blaCTX-M3a [18] and blaCTX-M-18 [19]) or nearly identical to the ancestors of plasmidic clades (Figure 1) have been found in the chromosomes of Kluyvera species, whereas many of the other plasmidic class A β-lactamases have been mobilized much longer, perhaps even since ancient times. In many cases, no chromosomal ancestor is identified and the plasmidic resistance genes are not closely related to the chromosomal resistance genes of any identified groups of bacteria.
Discussion
Although the use of antimicrobial agents generally has enhanced the spread of antimicrobial drug resistance among bacteria by providing the selective pressure needed for the emergence of novel resistance determinants, selective pressure alone does not explain the increasing frequency with which blaCTX-M alleles have been noted in bacterial populations in recent years (16,33). Although blaCTX-M alleles tend to be located on transmissible plasmids and transposable elements, which clearly facilitate their dissemination, the repeated mobilization of the blaCTX-Ms from the chromosomes principally among Kluyera species is most intriguing. The mechanistic basis underlying this repeated mobilization to plasmids remains unknown. Whether the chromosomes of Kluyvera species have some unique aspect that enhances the mobilization of the blaCTX-M genes remains to be determined. Other factors, such as exposure of the isolates to specific antimicrobial agents or to environmental changes that facilitate the mobilization of blaCTX-Ms to plasmids also need to be investigated.
Two insertion elements are known to contribute to the mobilization of blaCTX-Ms. The first, which is associated with the CTX-M-2 and CTX-M-9 groups, is ISCR1 (34) and the second, which is associated with the CTX-M-1, CTX-M-8, and CTX-M-25 groups is ISEcp1 (35). According to our phylogenetic analysis (Figure 1), 4 mobilizations can be attributed to each of these insertion type elements. Thus, both elements seem to promote equal frequencies of mobilization of blaCTX-Ms. Notably, ISCR1 was also reported to be responsible for the mobilization of both blaVEB and blaPER alleles, but neither of these resistance determinants has been reported to have an unusually high rate of mobilizations from chromosomal locations to plasmids.
Another factor that may contribute to the rate of mobilizations of the blaCTX-M resistance determinants is the frequency of plasmids in bacterial populations. As the number of plasmids increases in microbial populations, so does the number of target replicons. A comparison of the percentage of bacterial strains that contained plasmids in the pre–antimicrobial drug era (36) with the percentage of contemporary strains that carry plasmids (37,38) indicates that the frequency of plasmid carriage has increased from 19% to 58%–100%, depending on the species surveyed. Although the collection methods and resistance detection assays varied in the studies used for this comparison (which may have introduced biases toward an increasing frequency of plasmids), few doubt that plasmid carriage is much more common among bacterial strains in the antimicrobial drug era (39,40). Unfortunately, specific information about plasmid carriage of Kluyvera species versus other Enterobacteriaceae is not available.
Regardless of the mechanism, the increased number of mobilizations of blaCTX-Ms from their chromosomal locations among relatively rare human pathogens to plasmids that circulate widely among several important human and animal pathogens (particularly among E. coli) is a serious public health concern. The results of our study indicate the potential for an increase in the rate of mobilization of a variety of other resistance determinants to plasmids. Such an increase could result in more rapid mobilizations of novel resistance determinants and contribute to the accelerated spread of antimicrobial resistance determinants among a large spectrum of bacterial pathogens.
This study was supported as part of Phase V of Project ICARE through unrestricted research grants to the Rollins School of Public Health of Emory University from AstraZeneca, bioMérieux, Elan, J&J PRD, Pfizer, and 3M Health Care; and by start-up support from University of California, Merced.
Dr Barlow is a founding faculty member at the University of California, Merced. Her research focuses on the evolution of plasmidic resistance determinants, with particular emphasis on β-lactamases.
References
- Lawrence JG. Clustering of antibiotic resistance genes: beyond the selfish operon. American Society for Microbiology. ASM News. 2000;66:281–6 [cited 26 Jan 2008]. Available from http://newsarchive.asm.org/may00/feature2.asp
- Kim S, Kim J, Kang Y, Park Y, Lee B. Occurrence of extended-spectrum beta-lactamases in members of the genus Shigella in the Republic of Korea. J Clin Microbiol. 2004;42:5264–9.
- Baraniak A, Fiett J, Mrówka A, Walory J, Hryniewicz W, Gniadkowski M. Evolution of TEM-type extended-spectrum beta-lactamases in clinical Enterobacteriaceae strains in Poland. Antimicrob Agents Chemother. 2005;49:1872–80.
- Brinas L, Lantero M, de Diego I, Alvarez M, Zarazaga M, Torres C. Mechanisms of resistance to expanded-spectrum cephalosporins in Escherichia coli isolates recovered in a Spanish hospital. J Antimicrob Chemother. 2005;56:1107–10.
- Ho PL, Ho AY, Chow KH, Wong RC, Duan RS, Ho WL, et al. Occurrence and molecular analysis of extended-spectrum {beta}-lactamase-producing Proteus mirabilis in Hong Kong, 1999–2002. J Antimicrob Chemother. 2005;55:840–5.
- Ho PL, Shek RH, Chow KH, Duan RS, Mak GC, Lai EL, et al. Detection and characterization of extended-spectrum beta-lactamases among bloodstream isolates of Enterobacter spp. in Hong Kong, 2000–2002. J Antimicrob Chemother. 2005;55:326–32.
- Kruger T, Szabo D, Keddy KH, Deeley K, Marsh JW, Hujer AM, et al. Infections with nontyphoidal Salmonella species producing TEM-63 or a novel TEM enzyme, TEM-131, in South Africa. Antimicrob Agents Chemother. 2004;48:4263–70.
- Schlesinger J, Navon-Venezia S, Chmelnitsky I, Hammer-Münz O, Leavitt A, Gold HS, et al. Extended-spectrum beta-lactamases among Enterobacter isolates obtained in Tel Aviv, Israel. Antimicrob Agents Chemother. 2005;49:1150–6.
- Tasli H, Bahar IH. Molecular characterization of TEM- and SHV-derived extended-spectrum beta-lactamases in hospital-based Enterobacteriaceae in Turkey. Jpn J Infect Dis. 2005;58:162–7.
- Medeiros AA. Evolution and dissemination of beta-lactamases accelerated by generations of beta-lactam antibiotics. Clin Infect Dis. 1997;24(Suppl 1):S19–45.
- Datta N, Kontomichalou P. Penicillinase synthesis controlled by infectious R factors in Enterobacteriacae. Nature. 1965;208:239–41.
- Barlow M, Hall BG. Predicting evolutionary potential: in vitro evolution accurately reproduces natural evolution of the TEM beta-lactamase. Genetics. 2002;160:823–32.
- Hall BG, Barlow M. Evolution of the serine beta-lactamases: past, present and future. Drug Resist Updat. 2004;7:111–23.
- Roupas A, Pitton JS. R factor-mediated and chromosomal resistance to ampicillin in Escherichia coli. Antimicrob Agents Chemother. 1974;5:186–91.
- Haeggman S, Löfdahl S, Paauw A, Verhoef J, Brisse S. Diversity and evolution of the class A chromosomal beta-lactamase gene in Klebsiella pneumoniae. Antimicrob Agents Chemother. 2004;48:2400–8.
- Bonnet R. Growing group of extended-spectrum beta-lactamases: the CTX-M enzymes. Antimicrob Agents Chemother. 2004;48:1–14.
- Bauernfeind A, Grimm H, Schweighart S. A new plasmidic cefotaximase in a clinical isolate of Escherichia coli. Infection. 1990;18:294–8.
- Rodríguez MM, Power P, Radice M, Vay C, Famiglietti A, Galleni M, et al. Chromosome-encoded CTX-M-3 from Kluyvera ascorbata: a possible origin of plasmid-borne CTX-M-1-derived cefotaximases. Antimicrob Agents Chemother. 2004;48:4895–7.
- Boyd DA, Olson AB, Silverman M, McGeer A, Willey BM, Pong-Porter V, et al. Identification of a progenitor of the CTX-M-9 group of extended spectrum beta-lactamases from Kluyvera spp. isolated in Guyana. In: 44th Interscience Conference on Antimicrobial Agents and Chemotherapy. 2004 Oct 30–Nov 2. Washington: American Society for Microbiology; 2004.
- Poirel L, Kampfer P, Nordmann P. Chromosome-encoded Ambler class A beta-lactamase of Kluyvera georgiana, a probable progenitor of a subgroup of CTX-M extended-spectrum beta-lactamases. Antimicrob Agents Chemother. 2002;46:4038–40.
- Humeniuk C, Arlet G, Gautier V, Grimont P, Labia R, Philippon A. Beta-lactamases of Kluyvera ascorbata, probable progenitors of some plasmid-encoded CTX-M types. Antimicrob Agents Chemother. 2002;46:3045–9.
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10.
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–402.
- Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The ClustalX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–82.
- Yang Z. Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Mol Biol Evol. 1998;15:568–73.
- Huelsenbeck JP, Ronquist F. MrBayes: Bayesian inference of phylogenetic analysis. Bioinformatics. 2001;17:754–5.
- Tavaré L. Some probabilistic and statistical problems on the analysis of DNA sequences. Lec Math Life Sci. 1986;17:57–86.
- Swofford DL. PAUP*. Phylogenetic analysis using parsimony (*and other methods). Sunderland (MA): Sinauer Associates; 2000.
- Bauernfeind A, Casellas JM, Goldberg M, Holley M, Jungwirth R, Mangold P, et al. A new plasmidic cefotaximase from patients infected with Salmonella typhimurium. Infection. 1992;20:158–63.
- Olson AB, Silverman M, Boyd DA, McGeer A, Willey BM, Pong-Porter V, et al. Identification of a progenitor of the CTX-M-9 group of extended-spectrum beta-lactamases from Kluyvera georgiana isolated in Guyana. Antimicrob Agents Chemother. 2005;49:2112–5.
- Baraniak A, Sadowy E, Hryniewicz W, Gniadkowski M. Two different extended-spectrum beta-lactamases (ESBLs) in one of the first ESBL-producing salmonella isolates in Poland. J Clin Microbiol. 2002;40:1095–7.
- Ford PJ, Avison MB. Evolutionary mapping of the SHV beta-lactamase and evidence for two separate IS26-dependent blaSHV mobilization events from the Klebsiella pneumoniae chromosome. J Antimicrob Chemother. 2004;54:69–75.
- Rodríguez-Baño J, Navarro MD, Romero L, Muniain MA, de Cueto M, Ríos MJ, et al. Bacteremia due to extended-spectrum beta -lactamase-producing Escherichia coli in the CTX-M era: a new clinical challenge. Clin Infect Dis. 2006;43:1407–14.
- Toleman MA, Bennett PM, Walsh TR. ISCR elements: novel gene-capturing systems of the 21st century? Microbiol Mol Biol Rev. 2006;70:296–316.
- Poirel L, Lartigue MF, Decousser JW, Nordmann P. ISEcp1B-mediated transposition of blaCTX-M in Escherichia coli. Antimicrob Agents Chemother. 2005;49:447–50.
- Datta N, Hughes VM. Plasmids of the same Inc groups in Enterobacteria before and after the medical use of antibiotics. Nature. 1983;306:616–7.
- Souza V, Rocha M, Valera A, Eguiarte LE. Genetic structure of natural populations of Escherichia coli in wild hosts on different continents. Appl Environ Microbiol. 1999;65:3373–85.
- Wan KF, Radu S, Cheah YK, Benjamin PG, Ling CM, Hon SF, et al. Antibiotic resistance, plasmid profile and RAPD-PCR analysis of enteropathogenic Escherichia coli (EPEC) clinical isolates. Southeast Asian J Trop Med Public Health. 2003;34:620–6.
- Rainey PB, Cooper TF. Evolution of bacterial diversity and the origins of modularity. Res Microbiol. 2004;155:370–5.
- Mirkin BG, Fenner TI, Galperin MY, Koonin EV. Algorithms for computing parsimonious evolutionary scenarios for genome evolution, the last universal common ancestor and dominance of horizontal gene transfer in the evolution of prokaryotes. BMC Evol Biol. 2003;3:2.
Figures
Figure 1. Phylogenetic analysis of blaCTX-Ms.
This tree was calculated by Bayesian inference. Number of mutations occurring
along each branch are given along the length of the branch...
Figure 2. Phylogenetic analysis of class A β-lactamases
calculated by Bayesian inference. Number of mutations occurring along each
branch are represented visually by the lengths of the branches...
Tables
Table. Average distances from root and dn/ds ratios of monophyletic clades
Appendix Table. Taxon names, organisms, and GenBank accession numbers
Suggested Citation for this Article
Barlow M, Reik RA, Jacobs SD, Medina M, Meyer MP, McGowan JE Jr, et al. High rate of mobilization for blaCTX-Ms. Emerg Infect Dis [serial on the Internet]. 2008 Mar [date cited]. Available from http://www.cdc.gov/EID/content/14/3/423.htm
Please use the form below to submit correspondence to the authors or contact them at the following address:
Miriam Barlow, University of California, Merced, PO Box 2039, Merced, CA 95344, USA: email: mbarlow@ucmerced.edu
Please contact the EID Editors at eideditor@cdc.gov
The opinions expressed by authors contributing to this journal do not necessarily reflect the opinions of the U.S. Department of Health and Human Services, the Public Health Service, the Centers for Disease Control and Prevention, or the authors' affiliated institutions. Use of trade names is for identification only and does not imply endorsement by any of the groups named above.
This page posted February 26, 2008
This page last reviewed February 26, 2008
Centers for Disease Control and Prevention, 1600 Clifton Rd, Atlanta, GA 30333, U.S.A
Tel: (404) 639-3311 / Public Inquiries: (404) 639-3534 / (800) 311-3435