|
| |
||
| |
|||||||||||||||||
|
|||||||||||||||||
|
|||||||||||||||||
|
EID Home | Ahead of Print | Past Issues | EID Search | Contact Us | Announcements | Suggested Citation | Submit Manuscript
|
|
Research Enterobacter cloacae Outbreak and Emergence of Quinolone Resistance Gene in Dutch HospitalArmand Paauw,* Suggested citation for this article
Multidrug-resistance among Enterobacteriaceae, including resistance to quinolones, is increasing. Although quinolone resistance is predominantly caused by chromosomal mutations, it may also result from a plasmid-encoded qnr-gene (1). The QnrA determinant, a 218–amino acid protein, protects DNA gyrase and topoisomerase IV from the inhibitory activity of quinolones (2). However, expression of qnrA alone is frequently insufficient to reach Clinical and Laboratory Standards Institute breakpoints for ciprofloxacin resistance. Since first identified in 1994 in the United States, qnrA-like genes have been sporadically identified in Enterobacteriaceae worldwide (3–9). At the end of 2002, an outbreak of aminoglycoside-resistant Enterobacter cloacae infections with variable susceptibility for ciprofloxacin was detected in the University Medical Center Utrecht (UMCU), the Netherlands, involving >80 patients (10). The first aim of this study was to test the hypothesis that the variable susceptibility to ciprofloxacin of the outbreak strain was associated with plasmid-mediated qnrA and if so, to characterize the gene's molecular background and determine its ability to transfer in vitro as well as in vivo. Maximum circumstantial evidence for horizontal transfer in vivo with the outbreak strain as donor would be obtained if the following observations were made: 1) different species or strains collected from the same patient harbored the same qnrA-encoding plasmid; 2) this same qnrA-encoding plasmid was not found in patients without an epidemiologic link to the outbreak. The second aim of this study was to determine to what extent the qnrA gene is an emerging resistance problem in our hospital. Materials and MethodsBacterial IsolatesA total of 1,167 isolates were tested for a qnrA gene. Group I consisted of 178 E. cloacae pulsed-field gel electrophoresis (PFGE) typed isolates obtained from January 2001 to August 2003 from 159 patients (10). Of these, 83 tobramycin-resistant isolates obtained from 83 patients belonged to 1 clonal lineage (cluster I, outbreak strain). Five of these patients also carried a tobramycin-susceptible variant of the clonal lineage (IA). The remaining 95 E. cloacae isolates contained 5 small clusters of 2 isolates each (III–VII), 1 cluster with 6 isolates (VIII), 1 cluster with 3 isolates (II), and 70 unique strains. Groups II and III consisted of aminoglycoside-resistant, gram-negative bacteria identified in the hospital database that were other than the outbreak strain; these bacteria were isolated from patients with an outbreak strain (group II) as well as from patients not involved in the outbreak but admitted in the same period (January 2001–August 2003) (group III). Aminoglycoside resistance was the selection criterion because the outbreak strain was aminoglycoside-resistant, and these isolates are stored routinely in our laboratory. Group IV consisted of 867 Enterobacteriaceae isolates comprising 8 different species collected from 3 different origins: 269 clinical isolates from UMCU (1994–2000), 514 isolates from 23 European hospitals (1997–1998), and 84 fecal screening isolates from 53 patients at admission at UMCU (2000) (11). Identification and Susceptibility TestingIdentification and susceptibility testing of isolates obtained through 2000 were performed by using the VITEK1 System with AMS R09.1 software (bioMérieux, Marcy-L'etoile, France); isolates obtained after 2000 were tested by using the Phoenix 100 Automated Microbiology System version V3.22 software (Becton Dickinson Biosciences, Sparks, MD, USA). For susceptibility testing, Clinical and Laboratory Standards Institute guidelines were used (12). In the conjugation experiments, MICs were determined by using Etest (AB Biodisk, Solna, Sweden). Genotyping and Characterization of β-LactamasesE. cloacae isolates were typed by PFGE. Citrobacter freundii, Escherichia coli, and Klebsiella pneumoniae were typed by PFGE and random amplified polymorphic DNA (13). To determine the kind of β-lactamases the outbreak strain expressed, isoelectric focusing (IEF) was performed with Phastgels (pH gradient 3–9) with the PhastSystem (Pharmacia AB, Uppsala, Sweden) (14). β-lactamases of isoelectric pH (pI) 5.6 (TEM-1), pI 7.6 (SHV-2A), and pI 8.2 (blaCTX-M-9) and a broad range pI calibration set (Amersham Biosciences, Little Chalfont, UK) were used. β-lactamases were detected with nitrocefin (Oxoid, Basingstoke, UK). Detecting and Characterizing Resistance GenesTarget DNA for polymerase chain reaction (PCR) assays was extracted by heating bacterial suspensions for 10 min at 95°C. qnrA, blaCTX-M, and aadB were detected by PCR with primers and annealing temperatures described in the Table. The outbreak strain carried an integron containing an aadB gene encoding aminoglycoside resistance (17). Primers were developed to detect aadB gene and the downstream 3´-conserved segment (CS) of the integron in the same PCR (aadB-3´CS). PCR assays were performed for 30 or 35 cycles. The AmpC PCR tests were performed as described earlier, except that a single PCR format was used (18). The blaCTX-M gene from E. cloacae 02-477 was sequenced by using CTX-M-9 group sequence primers (Table). The flanking regions of the qnrA gene and the blaCTX-M-9 gene were determined by using a PCR and DNA sequencing strategy based on the sequences from In7, In36, In37, In60, and an integron from E. coli O159 (5,9,19–22). To confirm that the gene cassettes were part of a complex integron with qnrA or blaCTX-M-9, we used the Expand Long Template PCR system (Roche, Woerden, the Netherlands) employed primers to amplify sequences between the qnrA or blaCTX-M-9 and the possible gene cassettes. All amplified products were (partly) sequenced for confirmation. Sequencing was performed with Qiagen Quick (Qiagen, Westburg b.v., Leusden, the Netherlands) purified PCR products by using the BigDye Terminator v1.1 Cycle Sequencing Ready Reaction Kit and a 3100 capillary DNA sequencer (Applied Biosystems, Nieuwerkerk a/d Yssel, the Netherlands). Conjugation ExperimentsFor conjugation experiments, an E. coli K12 and a tobramycin-susceptible clinical E. cloacae (03-702) isolate of PFGE cluster IA were used as recipients. An E. cloacae (02-477) belonging to PFGE cluster I was used as donor. Conjugation was performed as described (23). MacConkey agar plates containing tobramycin (8 μg/mL) were used for counter selection, and transconjugants were selected on colony form. Conjugation was confirmed by a qnrA-specific PCR. Secondly, transconjugant E. coli C02-477A was used as a donor for qnrA-negative E. cloacae 03-702 belonging to cluster IA. Transconjugants were selected by using 15 μg/mL ampicillin-clavulanic acid and 5 μg/mL tobramycin. Transconjugants were characterized as described above. Detecting Resistance Genes on Plasmid by Southern HybridizationPlasmids were isolated with the Qiagen Plasmid Maxi Kit (Qiagen). Plasmid DNA was separated on 1% PFGE agarose (Bio-Rad Laboratories, Richmond, CA , USA) in 0.5× Tris-borate-EDTA, 0.05 mmol/L thiourea buffer at 14°C in CHEF DR-II apparatus (Bio-Rad). Run time was 22 h with a voltage of 6 V/cm and a linearly ramped pulse time of 30 to 70 s. The DNA was blotted and hybridized. The probes were PCR amplification products obtained with primers used to detect aadB-3´-CS, blaCTX-M-9, and qnrA genes (Table). Products were labeled with the AlkPhosDirect Reaction Kit (Amersham Biosciences) and detected with CPD-Star (Amersham Biosciences). ResultsqnrA1 in Outbreak StrainFor 78 (94%) of the 83 E. cloacae isolates in cluster I (outbreak strain), the qnrA-specific PCR was positive. To confirm results from the PCR, 2 fragments were sequenced. The obtained sequences were identical to the published sequence of qnrA1 (GenBank accession no. AY070235). Susceptibility testing showed that 87% of the 83 outbreak isolates were resistant or intermediate resistant to ciprofloxacin (43% resistant, 43% intermediate resistant), 100% were resistant to tobramycin, 63% to gentamicin, 2% to amikacin, 100% to ceftriaxone, 12% to trimethoprim-sulfamethoxazole, and 0% to carbapenems. A total of 81 (98%) of the 83 isolates harbored an aadB containing integron. IEF showed the presence of a β-lactamase with a pI of ≈8.2, which suggested the presence of either an AmpC β-lactamase or a CTX-M type extended-spectrum β-lactamase. No AmpC-specific amplification products were obtained. Eighty-two (99%) of the 83 isolates harbored a blaCTX-M gene. DNA sequencing showed the presence of blaCTX-M-9.
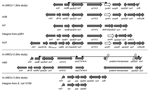
The plasmid (pQC) of conjugant E. coli C02-477A was isolated, and its size was estimated at 180 kb by agarose gel electrophoresis. Southern blotting that used specific probes confirmed that pQC contained the qnrA1 gene, the blaCTX-M-9 gene, and the integron with an aadB gene cassette (data not shown). Sequences flanking the qnrA1 and blaCTX-M-9 genes were comparable with 3 previously described class 1 integrons (Figure 1). The first integron (In-UMCU-1 accession no. AY987395), containing the qnrA1 gene, had the same additional structures as In36, orf513, qnrA1, ampR, plus a second copy of the 3´-conserved segment. The In36 integron contained the gene cassettes drf16 and aadA2, while In-UMCU-1 contained only the aadB gene cassette. In addition, the DNA sequences between the second sul1 gene and orf5 (bp 9606–9624 of In36) differed from the sequence of In-UMCU-1 (5). The second integron (In-UMCU-2, accession no. DQ108615), which contained blaCTX-M-9, was comparable to In60, but In60 contained the drf16 and aadA2 gene cassettes, while In-UMCU-2 contained the aadB gene cassette (21). The third integron (In-UMCU-3, accession no. DQ019420), which contained the gene cassettes sat, psp, and aadA2, was described previously in an enterotoxigenic E. coli O159 isolated in Japan (22). PCR amplification of the aadA2 gene of the donor, recipient, and transconjugants indicated that this third integron was also located on pQC. Evidence for Transfer of qnrA in vitroIn vitro conjugation experiments showed that pQC could be transferred both from and to the outbreak strain (Figure 2). pQC was successfully transferred from E. cloacae 02-477 to recipient E. coli K12. The resulting transconjugant E. coli was subsequently used as donor to transfer pQC to a type IA E. cloacae (03-702), which resulted in a successful transfer of pQC. pQC conferred increased ciprofloxacin MICs (from 6- to 10-fold) and resistance to tobramycin, tetracycline, and ceftriaxone to the transconjugants (Figure 2). Acquisition and loss of pQC were associated with 2 changes in the PFGE pattern. Evidence for Transfer of qnrA1 in vivoDifferent species or strains collected from the same patient harbored the same pQC. From 22 of the 53 patients with an outbreak strain, 35 other tobramycin-resistant, gram-negative clinical isolates were available. Eleven different strains obtained from 11 patients were positive for qnrA1, blaCTX-M-9, and aadB-3´-CS. These comprised 4 different species: C. freundii (n = 1), Enterobacter aerogenes (n = 1), E. coli (n = 7), and K. pneumoniae (n = 2). Plasmid isolation from 6 E. coli and 1 K. pneumoniae yielded a plasmid of the same size as the pQC in the outbreak strain. Because of its large size and possibly a very low copy number, only small amounts of plasmid DNA could be isolated. These amounts were insufficient to perform further comparative analyses by restriction fragment analysis or Southern blotting. Some E. cloacae strains with a strong epidemiologic link to the outbreak strain were also pQC positive. All isolates belonging to clusters III, VII, and VIII contained pQC as well as 5 E. cloacae isolates with a unique genotype. Plasmid isolation of 3 strains again showed a plasmid of the same size as the outbreak pQC. Three of the 5 unique isolates were obtained from patients who also harbored the outbreak strain. The qnrA gene, the aadB-containing integron, and the blaCTX-M-9 could not be detected in PFGE cluster IA, which is closely related to the outbreak strain (Figure 3). The loss of these genes was associated with increased susceptibility to ciprofloxacin, tobramycin, ceftriaxone, and tetracycline. In addition, an identical change in the PFGE pattern was observed, as in the in vitro experiments. These results suggest that the host may lose pQC in vivo. qnrA1 Recent Emergence as Clinical ProblempQC was not found in isolates obtained from patients without an epidemiologic link to the outbreak. No qnrA1 gene was detected in any of 83 aminoglycoside-resistant gram-negative organisms (44 E. coli, 19 K. pneumoniae, 4 Proteus mirabilis, 2 Klebsiella oxytoca, 2 E. cloacae, 1 Enterobacter sp., 7 C. freundii, 4 Serratia marcescens) obtained from 74 patients admitted to wards not involved in the outbreak during the outbreak period. Neither was qnrA1 detected in any of the 269 UMCU isolates or the 84 community isolates. Only 1 qnrA1-positive isolate was found in the 514 European isolates. This qnrA1-positive isolate was an E. cloacae organism isolated in 1999 at a surgical ward at UMCU that belonged to cluster III. The other 2 cluster III isolates were isolated at the same surgical ward during the outbreak period. DiscussionWe report a nosocomial outbreak with an R-plasmid–encoded qnrA1 gene. This plasmid (pQC) was first detected in an E. cloacae isolated in 1999 and subsequently in another E. cloacae strain that caused a large outbreak in our hospital, starting in 2001. Strong evidence is provided that this outbreak strain was the source from which pQC disseminated to other strains of the same species and other species by horizontal gene transfer. The qnrA1 gene was not detected in any of the hospital isolates (1994–2003) tested without an epidemiologic link to the outbreak strain, indicating that qnrA1 is a new emerging resistance trait in our hospital. pQC contained 3 different class 1 integrons. One integron was identical to an integron detected in an E. coli O159 from Japan (22). The 2 other integrons were complex integrons, In-UMCU-1 and In-UMCU-2, which were not described previously. Complex integrons are composed of a 5´-CS, gene cassettes, 3´-CS, qacΔE, sulI, additional genes, qacΔE, and sulI. These additional genes differ from gene cassettes by lacking a 59-bp element and having their own promoter (24). The qnrA1 gene in In-UMCU-1 was also present as an additional gene, as was the case for the 3 previous characterized qnrA1 genes in In36, In37, and the complex integron of pQR1 (5,9). The sequences of these genes were identical for In-UMCU-1, In 36, and In37, and slightly different for the integron on pQR1. The gene cassette content of the 4 integrons, however, was different, although all 4 possessed a gene encoding aminoglycoside resistance. All qnrA1-positive isolates reported in the literature also show resistance to cephalosporins (1,4–9,25–27). Therefore, qnrA1 seems to be closely associated with resistance to cephalosporins and aminoglycosides. How these comparable but different complex integrons arose is unclear. Either the same additional genes became associated with different integrons or the gene cassettes in an original complex integron were exchanged. Our study confirmed previous findings that the presence of qnrA1 does not necessarily lead to MICs above Clinical and Laboratory Standards Institute breakpoints for resistance to ciprofloxacin (1,3,7,25). Therefore, the presence of qnrA1 had no therapeutic consequences for the patients from whom these isolates were obtained. However, the increased MIC may provide the host bacterium a selective advantage in an environment of low concentrations of quinolones, increasing the bacterial numbers and therefore the absolute chance of a chromosomal mutation encoding resistance (7,25). The presence of a qnrA-carrying plasmid might even enhance the mutation rate encoding quinolone resistance (1). Furthermore, acquisition of qnrA by a host bacterium that already contains quinolone resistance mechanisms may raise MICs above the LCSI breakpoints (25,28,29). As shown in this study, the same plasmid may cause fluctuation in susceptibility in MICs in different recipients because of variation in porin expression or mutations in the gyrase or efflux pump-encoding genes (2). In conclusion, in a hospital setting the qnrA gene is advantageous for the host bacterium. Because of this gene's location on promiscuous R-plasmids, it is likely to emerge worldwide. Acknowledgments
References
Suggested citation
for this article: |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
||||||||||||
|
||||||||||||
|
|
|
EID Home | Top of Page | Ahead-of-Print | Past Issues | Suggested Citation | EID Search | Contact Us | Accessibility | Privacy Policy Notice | CDC Home | CDC Search | Health Topics A-Z |
||
|
This page
posted April 4, 2006 |
||
|
Emerging
Infectious Diseases Journal |
||