|
 |
 |
|
 | |||||
| |||||||||
American Society of Clinical Oncology/FDA Lung Cancer Endpoints Workshop April 15, 2003 [The following comments reflect the opinions of the workshop participants and do not necessarily represent the views of the FDA] Introductory Remarks Dr. Paul Bunn welcomed everyone in attendance and noted that the ASCO/FDA Lung Cancer Endpoints Panel had intentionally been established with a broad membership―including representatives from industry, advocacy groups, and the National Cancer Institute (NCI), as well as FDA and ASCO―in order to obtain input from a wide variety of perspectives on the topic of endpoints in clinical trials whose purpose is to support applications for the approval of new anti-cancer drugs or supplemental indications for already-approved anti-cancer drugs. All panel members share an interest in getting safe and effective drugs for cancer treatment to the public as quickly as possible, he said. In the past, Dr. Bunn continued, most anti-cancer drugs have been cytotoxic and survival has been the primary endpoint of principal interest in clinical trials of new cancer therapies. He commended FDA for recognizing the need to consider other primary endpoints now that a wider range of anti-cancer drugs are in development. Dr. Pazdur noted that, by statute, FDA can take advice related to oncologic drugs only from the Oncology Drugs Advisory Committee (ODAC). The purpose of this meeting was to have a wide-ranging discussion about the positive and negative aspects of various endpoints for trials of drugs to treat lung cancer in advance of consideration of the topic by ODAC. The discussions at this meeting, together with those at later meetings that will focus on endpoints for trials of drugs to treat other forms of cancer, would form a basis for a report to ODAC. No part of the discussion would focus on any specific drug that has either been approved or is under review at FDA. Endpoints for trials of chemopreventive drugs will be considered in another forum, Dr. Pazdur said. Because of the range of issues to be considered, an adequate discussion of endpoints for both treatment and chemoprevention trials during a one-day meeting was regarded as impractical. Regulatory Background: Standards and Endpoints for Drug Approvals (Dr. Grant Williams, FDA) General Requirements for Drug Approval Dr. Williams briefly reviewed the historical background to FDA's involvement in the drug approval process. The agency became responsible for evaluating the safety of drugs in 1933. In 1962, the Food, Drug, and Cosmetic Act was amended to require FDA to ensure that all drugs demonstrate efficacy "in adequate and well controlled studies" prior to marketing. There are two routes to the approval of a new drug in the United States: regular approval, which requires the demonstration of clinical benefit or an established surrogate for clinical benefit, and accelerated approval, which can be based on a surrogate endpoint that is considered "reasonably likely to predict clinical benefit." Regular Approval Section 505(d) of the Food, Drug, and Cosmetic Act defines "substantial evidence" of efficacy as "adequate and well controlled investigations." FDA has generally interpreted this to mean that multiple trials are required. However, in some cases, drugs have been approved on the basis of a single trial. FDA's 1998 efficacy guidance document states that a single trial is acceptable "generally only in cases in which a single multicenter study of excellent design provided highly reliable and statistically strong evidence of an important clinical benefit... and a confirmatory study would have been difficult to conduct on ethical grounds." A single trial may be considered adequate for approval of a supplemental indication for an already approved drug. Since the mid-1980s, FDA's policy has been to grant regular drug approvals on the basis of a demonstrated improvement in survival, tumor-related symptoms, or disease-free survival(in selected settings). To date FDA has not granted approval to a drug on the basis of an improvement in global quality of life. In the early 1990s, a joint FDA/NCI white paper recommended that complete response rate (in settings such as acute leukemia), and partial response rate (in settings such as hormonal therapy for breast cancer) be regarded as "established surrogates" for clinical benefit that could be used to support regular approval. FDA staff recently reviewed the endpoints used between 1990 and 2002 to approve new cancer-drug applications.1 Endpoints other than survival were the basis for 73% (48/66) of all drug approvals and for 67% (37/55) of all regular approvals (Table 1). Table 2 provides examples of oncology drugs approved on the basis of endpoints other than survival. TABLE 1. Summary of Endpoints for Regular Approval of Oncology Drug Marketing Applications, 1/1/90 to 11/1/02
Derived from Johnson et al. J Clin Oncol 2003;21(7):1404-1411 RR response rate TTP time to progression DFS disease-free survival TABLE 2: Examples of Oncology Drugs Approved on the Basis of Endpoints Other Than Survival
Accelerated Approval Accelerated approval (AA) allows approval on a less-established surrogate end point, one that is reasonably likely to predict clinical benefit. This approval mechanism may only be used when the drug provides a benefit over available therapy. Post-marketing (Phase 4) studies are required to verify the predicted clinical benefit.
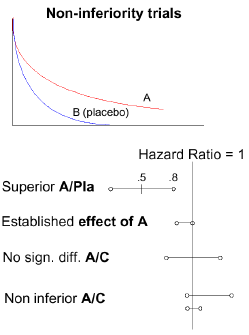
The surrogate end point results must represent substantial evidence from well-controlled studies. A trend toward improved survival, with no other evidence of a benefit, is an insufficient basis for accelerated approval. On the other hand, AA has been granted based on tumor response rates from single-arm studies. In the refractory setting, where there is no available therapy, single-arm studies can provide substantial evidence of response rates that are better than available therapy and are reasonably likely to predict benefit. To date, FDA has granted AA to 19 New Drug Applications (NDAs) or Biological License Applications (BLAs), involving 16 drugs, for new treatment indications in oncology. ODAC reviewed the AA experience at its March 2003 meeting. ODAC recommended that sponsors discuss Phase 4 confirmatory studies early with the FDA and incorporate them into the drug development plan. ODAC wanted to be consulted during discussion of phase 4 trial designs. Single-arm trials are the quickest way to obtain accelerated approval because they require the fewest patients. However, a single-arm trial can show benefit over available therapy only if no available therapy is effective. Thus, a single-arm trial could be used to support approval only for treatment of refractory disease. Additionally, a single-arm trial provides only limited ability to evaluate valuable endpoints such as survival, time to progression (TTP), and quality of life (QOL). Randomized trials require a larger number of patients and a longer period of time to complete. A randomized trial, however, may support accelerated approval at any disease stage if the surrogate endpoint is shown to be "reasonably likely" to improve on available therapy. A randomized trial permits the use of an add-on design (A vs A + B) and the use of a variety of endpoints, such as TTP, survival, and endpoints that require blinding (e.g., symptoms, QOL). Additionally, a three-arm randomized trial can define an individual drug's contribution to the treatment effect; an example of this was the trial of oxaliplatin vs. 5-fluorouracil (5FU)/leucovorin (LCV) vs. oxaliplatin plus 5FU/LCV in patients with advanced colorectal cancer. This study showed and advantage in both response rate and TTP for the combination arm over the other arms and supported AA of oxaliplatin. General Issues Regarding Endpoints Survival. Survival is obviously the "gold standard" endpoint for clinical benefit. When a randomized trial(s) clearly demonstrates that an experimental drug improves survival compared with standard therapy, approval is likely. Crossover from the control arm to the experimental arm of the trial may complicate the ability to demonstrate a survival benefit. It is more difficult to assess efficacy when a sponsor claims that its experimental drug is as good as current standard therapy. FDA terms such trials "non-inferiority trials" rather than equivalency trials because it can never be statistically proven that two treatments are equivalent. Non-inferiority provide assurance that the new drug is not worse than the control drug by some prespecified amount (the non-inferiority margin). A critical issue in determining the non-inferiority margin is the strength of evidence that the standard drug is effective. (See figure below.) Suppose, for example, that the maker of a new cancer drug C claims it is as good as the standard drug A but has a toxicity advantage―it does not cause the patient's hair to fall out. Ten years ago, when drug A was approved, its survival curve showed nearly a 50% difference in median survival compared to placebo. However, the 95% confidence intervals only provide assurance that the hazard ratio is at least 0.8 compared to placebo.
Now a trial is done to compare drug A with the new drug C. In the first example, the hazard ratio is 1, but the confidence interval extends to 0.7, suggesting that there is a reasonable likelihood that drug C may be no better than placebo. In the next two cases, the confidence intervals provide assurance that some fraction of the treatment effect has been maintained. In the first case, the point estimate suggests that the new drug C is better than A, but not significantly so. Even with wide confidence intervals, retention of effect is assured. In the next example, the hazard ratio estimate is 1, and because a large number of patients were studied, the confidence intervals are narrow and retention of activity is assured. Numerous other issues must also be considered with non-inferiority trial designs, such as whether the populations in the historical and current data are similar and whether the trials were carefully performed to minimize data variation. An FDA working group is evaluating various methods of evaluating the results of such studies. At present, the agency feels more secure evaluating trials designed to demonstrate the superiority of one drug over another. Non-inferiority designs are particularly problematic when current lung cancer regimens are being compared. Tumor Response Rate. An advantage of using tumor response rate as an endpoint is that it can be assessed in single arm study. Response rate, however, documents activity in only a subset of patients, whereas toxicity is generally experienced by all patients. In some settings, response rate has been considered to be an established surrogate. When topotecan was evaluated as a single agent for the treatment of refractory small-cell lung cancer (SCLC), ODAC determined that response rate was a reasonably likely surrogate for clinical benefit in that setting. Time to Progression. Critical regulatory questions concerning the use of time to progression (TTP) as an endpoint are whether it measures clinical benefit and whether it is reliable. The use of TTP has several advantages: TTP is measured in all patients and may therefore be a better measure of overall benefit than response rate. Because TTP does not require massive tumor shrinkage, it may be a better measure of benefit for cytostatic agents. From a practical standpoint, progression is often the basis for a change in therapy. Therefore, an advantage of using TTP rather than survival as an endpoint is that TTP is measured before patients change therapies or cross over. Because progression often occurs months to years before death, smaller studies are required to demonstrate improved TTP than are needed to show improved survival. Finally, some would argue that delaying progression has face validity as an indicator of clinical benefit because progression is a necessary step between cancer growth and patient morbidity and/or death. However, TTP is an indirect measure of clinical benefit. The clinical significance of small differences in TTP may be unclear, especially when one is evaluating toxic treatments. Careful assessment of progression at frequent intervals can be costly and labor-intensive. There are concerns about ascertainment bias in unblinded trials and questions about the reliability of small differences in TTP that are often observed in trials. One critical difference between analyses of survival and of TTP is that the date of death does not change regardless of censoring or the evaluation schedule. On the other hand, the date assigned for progression is usually the date of the next scheduled visit, which occurs some time after the actual date of progression and longer TTP is observed when a longer interval occurs between patient assessments. Bias can occur if follow-up schedules are not symmetric on the study arms. Tumor-Related Symptoms. FDA considers improvement in tumor-related symptoms to be a clinical benefit, not a surrogate for clinical benefit. Studies that adequately demonstrate improvement in tumor symptoms can support regular approval. Improvement in tumor-related symptoms have been important in the approval of several new oncology drugs (e.g. in settings of airway obstruction due to lung or esophageal cancer, cutaneous or subcutaneous tumors, and painful bone metastases). Impediments to the use of tumor-related symptoms as an endpoint include lack of blinding and missing data. Time to Symptomatic Progression. Time to symptomatic progression has been suggested as an endpoint, but, to date, has not been used for approval of a new cancer drug. A major problem with this end point would be loss of data when patients are withdrawn from the study due to objective progression. Discussion Dr. Bunn noted that in some instances only a single randomized trial may have been performed, but additional studies using historical controls may be deemed to be adequate and well controlled. For example, vinorelbine was approved both as a single agent and as part of a combination regimen to treat lung cancer on the basis of one randomized trial comparing it to 5FU and leucovorin and several single-arm Phase 2 studies. Dr. Saxman observed that the term "available therapy" is a confusing aspect of the guidelines on accelerated approval. Often there is no FDA-approved drug for an indication, although drugs may be used off-label and published studies support such off-label use. Dr. Williams responded that FDA's draft guidance states that a therapy is considered "available" if it is FDA-approved or if there is substantial evidence in the literature to support its use. Dr. Williams pointed out that the accelerated approval regulations do not allow a level of evidence about an established endpoint that is insufficient to support regular approval to be used to support accelerated approval. However, accelerated approval has been granted in situations in which clinical benefit is established but it is uncertain whether that benefit predicts an improvement in the ultimate outcome (e.g., whether disease-free survival at 3 years predicts disease-free survival at 5 years). A lengthy discussion took place about the difficulties of statistical interpretation of the results of non-inferiority trials. One major concern is whether historical estimates of the effect of the active comparator are appropriate. FDA statisticians have recognized the need for adjustment to account for estimate variability. Dr. Piantadosi suggested that some such adjustments may lead to overly conservative interpretation of results and underestimation of actual clinical benefit. However, solutions that have been proposed to address this problem introduce a greater degree of subjectivity into the interpretation of trial results. FDA has not, to date used Bayesian statistical methods to analyze phase 3 cancer trials, however FDA frequently discusses Bayesian methods (and is holding a workshop on the issue in about a month). Dr. Bunn commented that involving ODAC members in early discussions with sponsors about study design could be very helpful. In particular, the ODAC statistician could be involved in discussions about the design of non-inferiority trials. It was noted that for the last 18 months FDA has been inviting ODAC members to submit written critiques of the design of pivotal studies. Dr. Pazdur noted that most sponsors often seem unwilling to conduct more than one randomized trial. As a result, most New Drug Application submissions to FDA now include only one randomized trial. Thus, confirmation of a trial's results is lacking and provides less confidence about the estimate of the treatment effect. This presents a serious problem for non-inferiority trials in lung cancer. The discussion shifted to how bias in ascertaining the date of disease progression might be corrected. Dr. Bunn noted that average TTP in advanced lung cancer is 4 months; a 25% (or 1 month) improvement in the rate of recurrence or death is usually considered clinically relevant. One inherent problem in ascertaining the date of progression is that CT scans in pivotal trials are usually obtained during every other treatment cycle. Trial sponsors are reluctant to pay for CT scans during every cycle because this is not considered standard care. Different cycle lengths can also lead to confusion and bias. Apparent differences in TTP may be could be due to time-dependent ascertainment bias. Dr. Williams suggested one possible solution to these difficulties would be to evaluate disease status only once, at one standard time. The progression end point would be thus dichotomized, patients would have either progressed or not progressed at that one time. This would eliminate time-dependent ascertainment bias and decrease the cost of monitoring, but would likely also decrease the power of the study. Dr. Pazdur noted that the regulations governing accelerated approvals provide for accelerated withdrawal of a drug from the market if efficacy is not ultimately demonstrated in Phase 4 trials. In practice, however, it is extremely difficult to remove a drug from the market for lack of efficacy. FDA holds individual meetings with sponsors to review their plans for Phase 4 trials; the agency's goal is to ensure that these trials are part of a comprehensive drug development plan. Dr. Fleming commented that it is generally much more difficult to conduct randomized, controlled studies once a drug is on the market. To some extent, these challenges can be overcome if the sponsor has prepared a careful strategy for conducting a Phase 4 trial. However, sponsors may not feel any urgency about conducting Phase 4 trials if there are no consequences for not doing so. In the absence of clear criteria for accelerated withdrawal when the results of a Phase 4 trial are negative, accelerated approval is tantamount to regular approval. Advisers to FDA may be more willing to recommend accelerated approval of a drug if they are assured that post-marketing studies will be carried out and that the drug will be withdrawn if it is shown to be ineffective. Dr. Keegan noted that FDA has required the inclusion of negative trial findings in drug labeling and advertising. Endpoints for FDA Approvals of Lung Cancer Drugs (Dr. Martin Cohen, FDA) One single agent (vinorelbine) and four combination regimens (vinorelbine/cisplatin, gemcitabine/cisplatin, paclitaxel/cisplatin, and docetaxel/cisplatin) have been approved for first-line treatment of regionally advanced or metastatic non-small-cell lung cancer (NSCLC), Dr. Cohen said. Three approvals were based on a statistically significant improvement in survival, one on a non-inferiority analysis, and one on a statistically significant improvement in response rate and TTP, with a trend toward a survival advantage. In the second-line setting, only single-agent docetaxel has been approved. Approvals for First-Line Treatment. Single-agent vinorelbine was compared with 5FU/leucovorin. Median and 1-year survival were 30 weeks and 24%, respectively, for vinorelbine vs. 22 weeks and 16% for the comparator regimen (p=0.06). The vinorelbine/cisplatin combination regimen was studied in two trials. In the first trial, median and 1-year survival were 7.8 months and 38%, respectively, for the combination regimen vs. 6.2 months and 22% for cisplatin alone (p=0.01). In the second trial, navelbine/cisplatin was compared with navelbine alone and with vindesine/cisplatin. Median survival was 9.2 months for navelbine/cisplatin, 7.2 months for navelbine alone, and 7.4 months for vindesine/cisplatin. One-year survival was 35%, 30%, and 27%, respectively (p=0.05). Gemcitabine/cisplatin was also evaluated in two trials. In the first trial, median survival was 9.0 months for gemcitabine/cisplatin vs. 7.6 months for cisplatin alone (p=0.008). In the second trial, median survival was 8.7 months for gemcitabine/cisplatin vs. 7.0 months for etoposide/cisplatin (p=0.18). There were no differences in overall survival. Two paclitaxel/cisplatin regimens (135 mg/m2 [T135] and 250 mg/m2 [T250]) were compared with etoposide/cisplatin. Response rates were 23% for T135 and 25% for T250 vs. 12% for the comparator regimen. TTP was also superior for both T135 and T250. Survival differences were not statistically significant, although a trend favored T250 (p=0.08). Although not included in labeling, an analysis pooling the paclitaxel arms showed a statistically significant survival increase. In a non-inferiority analysis, docetaxel/cisplatin was shown to be non-inferior to navelbine/cisplatin; the regimen used in the third arm of the study, docetaxel/carboplatin, did not meet the non-inferiority standard. Approval for Second-Line Treatment. Docetaxel was evaluated in the second-line setting in two trials. In the first trial, the response rate for docetaxel was 5.5%. Median survival was 7.5 months for docetaxel compared with 4.6 months for best supportive care (p=0.01). In the second trial, the response rate was 5.7% for docetaxel vs. 0.8% for the investigator's choice of alternative regimen. Median survival was similar in both arms; however, 1-year survival favored docetaxel (30% vs 20%; p<0.05). Discussion Dr. Bunn noted that ODAC had recommended rejection of an application for approval of docetaxel for second-line therapy on the basis of results from two single-arm trials because of uncertainty about the adequacy of historical controls in this setting. The sponsor then conducted the randomized trials that were the basis for ultimate approval of docetaxel as second-line therapy. Dr. Canetta observed that the docetaxel approval was the first non-inferiority approval of a cancer drug in lung cancer. The fact that FDA approved it without consulting ODAC may have been precedent-setting. Dr. Williams briefly described the approval process for porfimer sodium. The drug was ultimately approved for two indications: pulmonary obstruction and peripheral microinvasive disease in patients who were not candidates for surgery. Despite missing data, it was eventually determined to be at least as effective as laser therapy. Dr. Bunn noted that this approval was largely based on patient-reported outcomes. In the only approval for small cell lung cancer (SCLC) in over 14 years, topotecan was compared with cyclophosphamide, doxorubicin, and vincristine for second-line therapy of SCLC, Dr. Williams said. Although missing data precluded a statistical analysis of patient reported outcome data, trends in favor of topotecan were observed. In addition, the topotecan response rate was was comparable to the response rate of the control arm, CAV. The committee felt that in the setting of refractory, rapidly-progressive disease such as SCLC, the observed response rate constituted clinical benefit. On the basis of the response rate and trends in patient reported outcomes, the topotecan received approval for second-line treatment of SCLC. A weakness of the topotecan trial design was that it failed to rigorously define patient-reported outcomes prospectively, Dr. Bunn noted. Although statistically significant reductions were seen in individual symptoms such as pain and dyspnea, these symptoms had not been prospectively defined as endpoints and they were not evaluated with a validated instrument. An additional problem was that because the trial was unblinded, bias in patient reports of symptoms could not be ruled out. The committee felt, however, that the patient-reported outcomes provided clinical validation for the objective response rate and may also have been swayed by the fact that a single agent performed as well as a combination regimen. Dr. Talcott commented that historically controlled trials are subject to many potentially troubling biases and that measurement of response rates is subject to measurement error. Dr. Keegan noted that in the setting of refractory disease, when no alternative therapy exists, comparison of response rate with a historical control in effect means comparison of the expected response rate with no treatment. Underpowered trials are a major problem, Dr. Pazdur said. Patient numbers are often calculated on the basis of how many can realistically be accrued rather than on how many patients are needed to show a treatment effect. The availability of data from a single randomized trial compounds the problem of lack of statistical power. One of the questions FDA is wrestling with is whether, to ensure that trials are adequately powered, sponsors should be asked power studies for survival data even when survival is not the primary. One reason that sponsors are only willing to do a single randomized trial is the high degree of investment risk associated with trials of cytotoxic drugs, Dr. Pazdur continued. Sponsors are usually willing to conduct more than one trial of a hormonal drug because they have greater confidence that the drug will ultimately be proven effective. Dr. Bunn commented that because therapies are now available that provide a modest survival benefit in advanced lung cancer, it may no longer be reasonable to design trials to detect a 33% reduction in hazard. However, trials designed to detect a 25% hazard reduction may need to accrue over 1,000 patients. Regulatory Approval Endpoints for Lung Cancer Adopted Internationally Renzo Canetta, M.D. Dr. Canetta discussed different approaches to regulatory approval endpoints adopted in the United States (US), in the European Union (EU), and in Japan (JPN) for non-small cell lung cancer (nsclc) and small cell lung cancer (sclc). In general terms, a clear separation of approvals specifically intended for nsclc (Table 1) and for sclc (Table 2) started occurring from the mid 80's on. Before that time, drugs were simply and generically approved for the treatment of lung or of bronchial carcinoma (Table 3). For both nsclc and sclc, the US has traditionally considered survival as the major endpoint of choice for all of its recent regulatory approvals (from 1986 on), with the sole exception of porfimer sodium (approved on response rate) and intrapleural bleomycin (approved on lack of fluid accumulation recurrence). Of note, at least up to this point, no drug for the treatment of nsclc or of sclc has been approved in the US under the "Accelerated Approval" (Subpart H) regulation, utilizing surrogate endpoints of clinical benefit. In Europe, before the creation of the European Medicines Evaluation Agency (EMEA), a wide array of endpoints has been adopted by the EU Member States that eventually adhered to EMEA. These endpoints included response rate, time to progression, quality of life, and overall survival (the latter, in one case, as a result of a comprehensive meta-analysis of the literature on cisplatin in nsclc). Of interest, a recent guideline on the approval of anticancer agents issued by EMEA has indicated that time to progression can be evaluated as an endpoint for the approval of drugs for the treatment of metastatic disease. In Japan, the approach has traditionally been to accept objective (complete and partial) response rates in excess of 20%, with independent committee assessment of such responses. This level of activity has been required of single-agents in non-randomized trials (Table 4). This approach has been upheld until very recently, with the approvals of gefitinib in June 2002 for nsclc (19% response rate, 27% in the subset of Japanese patients and 11% in the subset of non-Japanese patients) and of amrubicin in December 2002 for both nsclc (23% response rate) and sclc (76% response rate). The general approach in Japan has been to require large, prospective, randomized trials as a phase IV commitment after the initial approval (Table 5). These studies have utilized standard control arms that had been considered to produce a survival effect (initially the combination of vindesine and cisplatin which, in combination with mitomycin and radiation therapy, had been shown to improve survival when compared in a randomized trial to radiation therapy alone and, more recently, the combination of irinotecan and cisplatin). In conclusion, different methodological approaches have been adopted in different regions of the world in order to approve new drugs for the treatment of lung cancer, ranging from the more open approach adopted in Japan (which utilizes response rates) to the more methodologically stringent approach adopted in the US (which has historically utilized survival in most cases).
Discussion Dr. Bunn noted that the Medicare program has traditionally deemed reimbursable any drug used to treat an FDA-approved indication. However, the Center for Medicare and Medicaid Services (CMS), the federal agency that administers the Medicare program, appears to be revising this policy. To date, one drug approved by FDA has not yet been deemed reimbursable by CMS. Sam Turner, Esq., ASCO counsel, said that CMS has questioned whether all FDA-approved drugs provide clinical benefit. Dr. Williams noted that the standard for accelerated approval of a drug is that it is "reasonably likely" to provide clinical benefit. Dr. Pazdur noted that European regulatory agencies have been more favorable than FDA to the use of TTP as an endpoint. Dr. Canetta said that this is a fairly recent development; to date, no lung cancer approval has been based on TTP. The Japanese drug regulatory system has many unique features, Dr. Pazdur said, including a lack of infrastructure to perform randomized studies and a reluctance to accept foreign studies as relevant to the Japanese population. The Japanese medical care system is also very different from the U.S. system. For example, in Japan oncology drugs are commonly administered by primary care physicians, internists, and other non-oncology specialists. For this reason, the Japanese system tends to focuses more on establishing safety than on establishing efficacy. Dr. Pazdur said that FDA accepts data from trials conducted outside the U.S., provided that the trials are adequate and well-controlled and that secondary treatments and supportive care measures are equivalent to those that would be provided in trials conducted in the U.S. Overwhelming evidence indicates that pharmacogenomic differences between nationalities or ethnic groups are insignificant, said Dr. Cohen. Dr. Bunn observed that although carboplatin may be the most widely used drug in lung cancer treatment, it is not approved to treat lung cancer in the U.S. Many trials of carboplatin and irinotecan have demonstrated the equivalence of these drugs, he added, suggesting that a meta-analysis of these studies might be worthwhile. Dr. Williams commented that it would be necessary for a sponsor to request approval of carboplatin for lung cancer treatment and submit data on the safety and efficacy of the drug for that indication. In addition, trials could be done of combination regimens including carboplatin. Dr. Gralla commented that in some of the trials Dr. Canetta presented as examples, QOL analysis was conducted retrospectively using an instrument that has not been validated in lung cancer. Dr. Pazdur noted that international regulatory agencies do not routinely communicate with each other about specific applications. Given the global nature of drug development, it could be useful to invite representatives from other countries' regulatory agencies to attend or participate in future meetings that will consider endpoints. Dr. Bunn said that ASCO could write to the European Medicines Evaluation Agency, the Canadian Health Protection Branch, and the Japanese drug regulatory body inviting them to send representatives to future meetings. Classical Lung Cancer Endpoints (Dr. David Johnson) Dr. Johnson discussed three classical endpoints―objective response rate, TTP, and survival―and one novel endpoint that has been proposed, the percentage of patients with disease progression at a uniform time point. Most of his presentation dealt with metastatic (stage 4) NSCLC. Objective response rate is a well-defined and widely accepted endpoint. However, it does not correlate well with overall survival, which makes its use as a surrogate endpoint problematic. This problem might be overcome if stable disease is included in the response rate. There is evidence from the literature that a higher response rate correlates with a reduction in tumor-induced symptoms such as pain and dyspnea. Although the use of TTP as a surrogate endpoint has been proposed, TTP is poorly defined and unstandardized and its relationship to patient benefit is unknown. Analysis of data from patients with stage 4 disease who were enrolled in NCI-supported cooperative group trials does suggest, however, an approximate correlation between TTP and overall survival―that is, median survival is roughly twice the time to progression. Overall survival is the most widely accepted and most commonly used endpoint. It is easily determined and definitive. In a well-designed randomized trial, an observed survival benefit can be confidently attributed to the experimental therapy. Percent progression at a uniform time point has been suggested as an endpoint that could be measured earlier in the course of a trial. The association of this endpoint with patient benefit is not well defined. It use implies that patients with stable disease have a survival outcome similar to those with tumor regression. An analysis of data from a National Cancer Institute of Canada (NCIC) trial showed that patients who had progressive disease at a specified time point had inferior survival to patients who responded to treatment or whose disease was stable. Similar results were obtained in a retrospective analysis of data on nonprogression at various predetermined time points in five Eastern Cooperative Oncology Group (ECOG) trials. Consistently, nonprogression at a predetermined point predicted survival more accurately than either response rate or TTP. Discussion Dr. Fleming observed that many although biological factors are correlated with clinical outcomes, correlates are not necessarily valid surrogates for clinical benefit. Dr. Piantadosi expressed concern about the methodology used in the analyses described by Dr. Johnson. He said he would not find any quantity of such aggregate data from relatively small, highly selected studies to convincingly make the case for the utility of a surrogate endpoint. Measurements in individual patients that showed progression to be predictive of a later outcome would be more compelling. Such a measurement could be useful if measured in a standardized fashion and if it were shown to occur earlier and more frequently than death. Determining TTP is subject to many vagaries, Dr. Piantadosi continued, including how frequently the patient is assessed, what tests are applied, and whether follow-up is active or passive. Any intermediate outcome derived from TTP would be subject to the same vagaries, regardless of how or when it is measured. Panel members discussed whether progression at 6 weeks could be considered a reasonably likely surrogate for clinical benefit. Dr. Williams noted that although measuring TTP at a uniform time point cannot eliminate bias, it does have the advantage that bias is applied equally. Dr. Fleming noted that validation of any surrogate endpoint involves capturing its net effect on the endpoint of interest, which requires considerably more data than are necessary to validate the clinical endpoint itself. However, a reduction in TTP could be demonstrated in a much smaller trial than would be required to show a survival benefit. The utility of progression as a surrogate endpoint is questionable because the biological relevance of progression to survival is unclear, said Dr. Talcott. Dr. Bunn responded that, given the confounding effect of secondary treatments on the evaluation of differences in survival, progression could be as valid an endpoint as survival and an effect on progression could be demonstrated in a smaller study. One problem with surrogate endpoints is they will vary according to the agent being tested, said Dr. Kaplan. For example, an anti-angiogenic agent may not demonstrate the same activity at a given point in time as a cytotoxic agent. Dr. Piantadosi identified two issues that should not be confused: firstly, the utility of surrogate endpoints as a way of making trials shorter and simplifying the approval process and, secondly, the utility of these endpoints in making treatment comparisons. Although there may be circumstances in which a good surrogate might make trials more efficient and marginally shorter, the goal of trials should be to produce definitive evidence on which to base an approval. Earlier in the development process, however, progression might be a useful alternative to tumor shrinkage, which is of limited utility as a surrogate endpoint and is not helpful in evaluating targeted therapies and cytostatic agents that do not affect tumor size. Evaluation of these agents requires a surrogate that can show that disease progression has slowed even though tumor shrinkage has not occurred. Dr. Johnson suggested that the validity of percent progression at a uniform time point as a surrogate endpoint should be evaluated in another data set. Drs. Fleming and Piantadosi agreed that the crucial question is whether progression reliably predicts outcome in individual patients. Two questions need to be answered: · Is there is an association between an earlier event (i.e., progression) and a later event (i.e., survival)? · Does a treatment effect on the earlier event predict a treatment effect on the later event? The first question could be answered in a series of single-arm trials; the second can only be answered in randomized trials. Dr. Piantadosi proposed that a TTP analysis be introduced prospectively into a large, already-planned study. In conclusion, panel members agreed that time to progression has not yet been validated as a surrogate endpoint but is worthy of further investigation. Among the many issues that must be resolved is a need to agree on the best ways of measuring and evaluating TTP. Non-Classical Endpoints in Lung Cancer―Patient-Reported Outcomes (Dr. Richard Gralla) Dr. Gralla began by noting that the term patient-reported outcomes (PROs) embraces a larger universe than either QOL or symptoms. PROs may include measures of pain, toxicity, and other symptoms that may not be included in a global QOL measure. Comprehensive measures of health-related QOL may be considered a subset of PROs. Dr. Gralla added that his comments referred primarily to the evaluation of agents with anti-cancer effects. More than 90% of lung cancer patients report two or more disease-related symptoms at presentation. These are most commonly pulmonary problems such as cough and dyspnea and the systemic symptoms of fatigue, pain, and anorexia (Hollen) additionally, patients have high degrees of psychological distress (Hopwood). Consequently, in addition to survival and response outcomes, information about treatment effects on PROs is important. A meta-analysis of NSCLC studies performed between 1991 and 20012 showed that chemotherapy improved survival over supportive care. However, newer chemotherapy regimens have not shown superior survival relative to each other. Survival is an appropriate endpoint when a survival difference is considered likely. Response is considered a somewhat unreliable measurement that is clinically useful only if it correlates with survival, QOL, or PROs. QOL is an appropriate endpoint when survival differences are unlikely. There are several potential benefits to assessing PROs in addition to more traditional focused outcomes such as survival and response. First, it has been demonstrated that health care professionals may underestimate the subjective or palliative benefits associated with anti-cancer treatments, when compared with patient self-reports. In the recently reported MILE study, outcomes reported by patients differed markedly from those perceived by physicians and nurses. Second, response rates tend to underestimate patient-reported benefits. PROs can improve with less than major response to treatment and may even improve with stable disease. Third, these palliative outcomes can change rapidly in lung cancer patients and, when measured frequently (at least every 3-4 weeks), can create an accurate picture of the disease course. Other biomedical endpoints cannot consistently predict the balance between symptom improvement and toxicity or the effect of delayed progression that is summarized in many PRO measures. However, although many randomized controlled trials in patients with lung cancer have incorporated PRO measures, to date no drug has received regulatory approval for the treatment of lung cancer solely on the basis of PRO measures. The influence of baseline QOL on survival was prospectively evaluated in a multicenter study of 673 patients with NSCLC. Patients who rated their baseline QOL at above the median for the group survived longer than those who rated their baseline QOL at below the median for the group. Baseline QOL was the single most powerful prognostic indicator of survival. Two terms are commonly used in the evaluation of the palliative or subjective benefits of chemotherapy in patients with cancer. Clinical benefit has been defined in new-agent testing to include control of three common cancer-related problems: pain, weight loss, and performance status. Quality of life is a multidimensional measure that provides an overview of a patient's status. QOL generally is considered to encompass five dimensions: physical, functional, social, psychological, and spiritual. Clinical benefit is included in the physical and functional dimensions of QOL, which are most likely to be affected by experimental interventions. Thus, although all dimensions of quality of life are important, when evaluating a new treatment it may be relevant to focus on those aspects most likely to be influenced by the intervention. For this reason, the Lung Cancer Symptom Scale (LCSS) focuses on identifying changes in the physical and functional dimensions of QOL. The LCSS is one of three QOL instruments that have been validated in lung cancer. The other are the European Organization for Research and Treatment of Cancer Quality of Life Questionnaire (EORTC-QLQ-Q30) and the Functional Assessment of Cancer Therapy lung cancer subscale (FACT-L). All three of these instruments have undergone fairly extensive field testing and appear to have acceptable psychometric properties.3 All have been translated into multiple languages, including French, Spanish, and numerous other European and Asian languagesOther questionnaires used in lung cancer studies include the Rotterdam Symptom Checklist and the Hospital Anxiety and Depression scale. However, these instruments are not lung-cancer-specific and published reports of their psychometric validity are variable. A subscale of the FACT-L instrument was used to measure QOL in the IDEAL 2 randomized Phase II trial of gefitinib (Iressa) at two dose levels in patients with NSCLC Palliation was noted rapidly (generally within 7 to 10 days) when it occurred. Responding patients had greater symptom relief than those with stable or progressive disease (43% reported symptom improvement, 34% quality of life improvement).4 Analytical problems associated with uncontrolled trials evaluating survival are also found in studies examining PROs. Like survival, PROs are best evaluated when data from all patients entered into appropriately powered controlled studies are included in the analysis. In single-arm trials, the use of appropriate standard palliative measures (e.g., analgesics, antitussives, oxygen) can confound the evaluation of QOL, leading to an overestimation of the benefit attributable to the study agent. On the other hand, if only a partial tumor response can result in patient-reported symptom relief, a major response is likely to underestimate QOL benefits. Studies have shown that the drop-out rate from analyses is not random; patients who are the most symptomatic and have the lowest performance status at trial entry are the first to be lost to evaluation (largely due to poorer survival, but also to lower follow-up rates). Any trial in which missing data increases with deteriorating patient health will underestimate worsening PRO outcomes. This paradoxical appearance of improvement as a result of the attrition of sicker patients complicates the evaluation of patients in uncontrolled studies and can minimize differences in PRO outcomes between therapies in randomized, controlled trials (in which the arm with inferior survival loses more symptomatic patients earlier, thus lessening differences between the study arms). Adherence―that is, the willingness and or ability of an individual to complete a questionnaire at the time specified by the protocol―has been a problem. One analysis indicated that disease progression is the major reason for non-completion, with patients going off study without follow-up being requested for PRO endpoints (Hollen). However, the results of a recent mesothelioma study suggests that these difficulties can be overcome by prospectively emphasizing PROs. In this study, a brief training session was organized for all investigative and data management personnel on the methods and role of HRQOL evaluation. Baseline QOL data were included in the assessment of eligibility for randomization and the need for vigilance in the assessment of PRO endpoints was continually emphasized during the trial. As a result of these efforts, more than 90% of the planned weekly QOL assessments occurred over the initial six cycles of the trial, despite the difficult and progressive nature of mesothelioma. In conclusion, Dr. Gralla identified the following as critical elements to be considered in the design of trials that use PROs as endpoints: · Use of valid, feasible, reliable, sensitive instruments that are appropriate for the disease stage and that yield consistent results across socioeconomic status, literacy, and culture or language differences in the study population. · Clearly defined primary and secondary endpoints. Because available validated instruments have different features, care in the selection of the instrument is advised. Endpoints to be analyzed must be prospectively defined and attention must be paid to methods of handling (or, more importantly, avoiding) missing data. · Use of an appropriate control group for comparison of outcomes. When assessing symptom changes, data on concomitant interventions affecting these outcomes must be collected and, when possible, controlled. · Methods to assure compliance with protocol-specified PRO assessments, including an emphasis on the study's commitment to evaluating PRO endpoints. Adequate patient follow-up must be considered mandatory. This point must be made clear both to individual investigators and to patients as part of the consent process. · When feasible, blinding of interventions to minimize bias. Discussion Dr. Fleming commented that although it is an important insight that patients with poorer QOL are more likely to be lost to follow up, there is no evidence that QOL directly influences survival. Rather, the disease process influences both survival and QOL; disease progression results in poorer QOL as well as poorer survival. QOL is also an independent domain in that improved QOL is a desirable outcome whether or not it is correlated with survival. Dr. Talcott pointed out that QOL can also be used as a prognostic factor that correlates with function and as a factor that helps to characterize individuals within study populations. Because the disease process influences both QOL and survival, it is plausible that effective intervention against the disease process could beneficially influence both these endpoints, said Dr. Fleming. However, it would be incorrect to conclude that improved quality of life leads to improved survival. Studies are needed that establish whether or not an intervention results in a survival benefit and/or a QOL benefit. Dr. Fleming described four domains in which challenges to the validity of QOL analysis remain. � Measurability/feasibility. Some progress has been made in this domain, although missing data remain a challenge. Complete follow-up is necessary to preserve the integrity of randomization. Incomplete follow-up tends to occur because patients have dropped out of therapy, because their condition has deteriorated, or because they have died. Intensive procedures such as those described by Dr. Gralla can ensure more complete capture of data from patients who have deteriorated or dropped out. The best way to deal with missing data is to prevent it. When therapy stops or side effects occur, it remains important to assess outcomes. · Reproducibility/internal consistency. Some progress has also been made in this domain. � Sensitivity/specificity. Measures must be sensitive to meaningful influences of treatment on QOL. Blinding is critical to obtain the lack of bias that is critical to achieving sensitivity and specificity. However, many trials in oncology are not blinded because the treatments are delivered in very different ways or the toxicity profiles are very different and it is considered unethical to introduce toxicity in the control arm in order to achieve blinding. � Clinical relevance. There must be confidence that a treatment-induced change is clinically meaningful. How large must the change be and how long must it be sustained to be considered clinically relevant? All components of an aggregate measures may not be of equal clinical relevance. When a score is a weighted average of multiple subscores, it is necessary to establish with confidence that a change in the average reflects a change in clinically important domains. For example, an intervention may be shown to have a beneficial effect on dyspnea, but if that symptom is clinically important to only one in five patients, its overall clinical relevance is not significant. If a sponsor believes an intervention will be effective against dyspnea, it should test that hypothesis in a study conducted in patients with that symptom. A distinction must be made between confirmatory and exploratory analyses, Dr. Fleming added. Data from any study should always be fully explored, but the results of post-hoc analyses cannot be interpreted in the same way as results obtained for a prespecified primary endpoint. Finally, because results may be influenced by ancillary therapies and placebo effects, it is crucial that controlled studies be performed. Dr. Talcott observed that QOL measurements are "noisier" than others and that power calculations that are adequate for other measures may not be adequate for measuring QOL. He added that a great deal more effort needs to be made to make the results of QOL measurement accessible to end users. Missing data. Dr. Burke noted that an FDA working group is attempting to address the issue of missing data by developing techniques that facilitate the capture of patient data electronically through the use of personal digital assistants and other electronic devices. Compliance with paper diaries is traditionally poor; it is hoped that the use of electronic devices will facilitate more reliable ascertainment of PROs. Dr. Gralla said he believes there are ways to account for patients who die. The important variable is the patient's QOL during the period preceding death. Just as in the past agreement has been reached on how to interpret survival and response data, there is a need now to agree on ways to measure QOL. Dr. Keegan noted that most cancer studies collect data to the point where patients progress, at which point they are lost to follow-up. One way to convey to sponsors the importance of minimizing missing data would be to set a threshold for the amount of data needed over a defined period of time to constitute a valid assessment of this endpoint (e.g., 95% data collection for X months). Protocols and statistical plans should define 'missing data, detail how to handle deaths, and provide a detailed plan for utilizing deaths and missing data in the analysis. Dr. Gralla said that Wittes proposed in 1985 that less than 85% data collection in any domain (not specifically QOL) should be regarded as problematic. Single symptom vs. multiple symptoms. Dr. Pazdur wondered whether, for drug approval purposes, it would be better to design studies to measure a drug's effect on a single symptom. Dr. Gralla responded that because most anti-cancer drugs are not directed at specific symptoms, the benefits of treatment are more likely to be measured "across the board" rather than in diminution of a specific symptom. Dr. Williams said that FDA has accepted the concept of measuring the treatment effect on a symptom specified by the patient. Dr. Fleming commented that this would be appropriate when there is heterogeneity across patients as to which symptom domains are most important. Dr. Bunn observed that because most patients with advanced lung cancer have multiple symptoms, instruments need to be able to address the impact of treatment on multiple symptoms. Dr. Fleming said that if multiple domains are being measured and if treatment is influencing all identified domains equally, then a global measure that averages the treatment effect on all domains is valid. However, if treatment disproportionately affects one or two domains, averaging the treatment effect will present a misleading picture. Toxicity. Dr. Talcott asked whether it was necessary to distinguish between QOL and symptoms. A good QOL instrument is one that adequately addresses the clinical situation. In lung cancer, the reality is that most treatments are quite toxic. Dr. Williams said that FDA may need to distinguish between QOL and symptoms because it is necessary to establish the efficacy of a treatment before it can be approved. A drug cannot be approved solely on the basis of lower toxicity; it must also be shown to be efficacious. The relationship between therapeutic toxicity and QOL is complex. Dr. Bunn noted that there have been trials in lung cancer in which one chemotherapeutic agent was shown to be considerably less toxic than another although both were equally efficacious. However, global QOL analysis showed no difference between patients in the two groups. Dr. Gralla said that in a breast cancer study in Manitoba, Canada, patients who received the more toxic therapy has better QOL outcomes. He added that patients may assess their QOL differently depending on when they are asked the question (e.g., at treatment, immediately after treatment, or 2 weeks later). In addition, levels of symptom-related distress vary between patients. The advantage of a global QOL measure is that it allows patients to quantify the individual, subjective benefit that they have obtained from a given therapy. Influence of supportive therapies. It was noted that depression is common, although not universal, in patients with lung cancer. Some studies have suggested that patients with lung cancer whose depression is treated have less pain and anxiety. In one recent study, which was not lung-cancer-specific, patients who were mildly to moderately depressed benefited from treatment with selective serotonin reuptake inhibitors. Dr. Gralla commented that the use of analgesics and antidepressants can be confounding factors in supportive care, which is why controlled trials are necessary. He noted that the appropriate way to measure the impact of a therapy on pain is to look at whether the patient used more, less, or the same amount of pain medication. Dr. Gralla referred to an unblinded study by Moore, in which each decile of performance status correlated directly Validity of PRO endpoints in uncontrolled studies. Dr. Gralla said that uncontrolled studies can identify aspects worthy of evaluation in a controlled trial. Dr. Keegan said that single-arm studies might indicate where minimal tumor response correlates with symptomatic improvement. Dr. Bunn said that in his opinion there are insufficient historical data to justify the use of QOL/PROs as an endpoint in uncontrolled trials. He referred to Tannock's review of double-blinded, placebo-controlled trials in advanced cancer. Both objective and subjective responses were considered. For objective response and placebo, the range was 0-7 (median ~2, 95% CI ~0-10). For subjective relief of symptoms, the placebo group had a range of 0-14 (median ~7, 95% CI 0-20). Validity of PROs as primary endpoints in Phase 3 trials. Dr. Burke observed that in drug approval submissions to FDA, PRO outcomes are frequently not well studied or well integrated into the protocol; sponsors frequently attach these endpoints as an appendix. The panel discussed at length what level of evidence would be considered adequate to demonstrate a clinical benefit based on PROs. The first example presented was that of a controlled, non-blinded trial of adequate size in which the primary endpoint is symptom improvement; the survival advantage for the experimental agent is insignificant; 90% of the data are collected; and a prespecified improvement is shown and confirmed in a second controlled trial. Dr. Talcott suggested that, at a minimum, sponsors must demonstrate the following: · Use of valid, relevant, comprehensive measures. · A plan for addressing missing data (i.e., sensitivity analysis). · Biological correlates that support efficacy. · No evidence of offsetting toxicity. Need to demonstrate efficacy. Dr. Williams reiterated that demonstrating a better toxicity profile that a comparator is insufficient for approval; it must also be demonstrated that the experimental drug has anti-tumor efficacy. Otherwise, an improvement in PROs could be an artifact of lower toxicity rather than a result of a tumor-related effect. Dr. Keegan noted that the approval of mitoxantrone was based on evidence that patients had improvement in bone pain symptoms that correlated with tumor-related effects. In this instance, FDA was confident that the symptomatic improvement was due to an effect on the tumor and not to a reduction in toxicity. Dr. Gralla asked whether an experimental drug that had demonstrated anti-cancer efficacy but a minimal survival advantage would be approvable on the basis of patient reports that it was easier to take than the approved regimen. Dr. Williams said the non-inferiority standard must be applied to show that efficacy has not been lost. Dr. Gralla commented that the non-inferiority standard is so conservative it verges on requiring a demonstration of superiority. Dr. Fleming pointed out that a reduction in toxicity may be attained by reducing the dose of an active therapy, thereby improving QOL. There must be confidence that a QOL benefit (possibly mediated through reduced toxicity) is achieved without giving up efficacy. If an intervention is favorably influencing QOL by means of a positive effect on the disease process, there is probably a trend toward improved survival, although it may not reach statistical significance. The use of a reasonable margin for non-inferiority will rule out the possibility that the QOL benefit was achieved at the expense of a survival benefit. In an unblinded trial, greater strength of evidence would be required to overcome uncertainty about the extent to which bias was introduced by the lack of blinding. Dr. Keegan said that even if PROs were the primary endpoint, FDA would want to see data showing that survival is not worse with the experimental agent. Dr. Pazdur noted, however, that symptom improvement is a primary clinical benefit that meets the criteria for drug approval; the law does not require demonstration of a survival benefit. PROs could be the basis for approval if they provide evidence of clinical benefit. The question is whether the instruments used to measure symptom improvement are reliable and reproducible. Instrument validity. Dr. Gralla stated that the strength of evidence for the three QOL instruments validated in lung cancer (LCSS, FACT-L, and EORTC-QLQ-Q30) is far greater than that supporting the NCI's common toxicity criteria. In response to an earlier comment by Dr. Burke that the validation process for an endpoint is treatment-specific and patient-population-specific, Dr. Gralla said it should not be necessary to re-establish instrument validity in every trial, adding that this is not required for other endpoints (e.g., CAT scan results, PSA values). Valid endpoints in adjuvant and neoadjuvant settings. Dr. Bunn noted that time to progression or recurrence has been the basis for drug approvals in melanoma. It is well documented in the literature that in most patients with advanced lung cancer, recurrence is diagnosed by symptomatic disease and that patients benefit from a delay in disease progression. Dr. Williams said that disease-free survival (DFS) has been an acceptable endpoint in the past, primarily in trials of breast cancer treatments. It was noted, however, that because most treatments for advanced lung cancer are toxic, there is a need to weigh treatment-related symptoms against disease-related symptoms. Dr. Bunn observed that about 20% of lung cancer patients die of co-morbid disease; therefore, extending a trial for a year in order to observe a survival effect will result in the loss of more patients due to co-morbid conditions. Dr. Fleming said that in the advanced disease setting, an intervention that substantially reduces the rate of progression may influence survival, but the effect on survival will be weaker. The advantage of using TTP as an endpoint is that progression is seen sooner than death. However, the fact that the effect on survival tends to be weaker than the effect on TTP affects sample size. Dr. Saxman said that very few randomized trials in lung cancer have shown a survival advantage in the adjuvant setting and that disease-free survival does not necessarily track with overall survival. Even when it does, the question is how much therapeutic toxicity is induced to lengthen DFS. Evaluating combinations of experimental agents. Dr. Canetta commented that targeted therapies are effective for tumors that depend on a sole pathway that can be blocked with a targeted agent (the Gleevec model). However, when there are parallel disease pathways, more than one experimental agent may be required to block them. Suppose, he suggested, that A and B are both experimental agents and that A+B shows a survival advantage over the standard combination regimen C+D. He asked whether this would be sufficient for approval of both A and B. Dr. Pazdur said that by law, FDA issues a licensing agreement for a specific drug and therefore must be satisfied that the drug (not a combination of drugs) is safe and effective. In a trial of A+B, it is possible that the survival advantage could be entirely due to one drug and that other drug is not effective. A three-arm trial (A vs. B vs. A+B) would provide the strongest evidence that the combination was necessary to produce the survival benefit. Preclinical data alone may be insufficient given the tenuous relationship between preclinical and clinical efficacy in oncology. Dr. Bunn posed a slightly different hypothetical scenario: Suppose, he said, that phase 2 data show that neither A nor B alone is effective but that A+B is very effective. Dr. Pazdur said that whether such data would be sufficient for approval would depend on the magnitude of the response and the quality of the trial and on whether or not the results had been duplicated. He emphasized that science must precede regulatory action. FDA has a statutory responsibility to the patients of the United States to ensure that approved drugs are safe and effective. FDA would consider a submission for approval of a drug for treatment of patients with a specific genetic abnormality provided that the scientific rationale was sound, Dr. Pazdur added. An advantage of such an approach could be that a greater treatment effect could be seen with a smaller sample size. Dr. Burke said FDA is very willing to advise sponsors early in the planning process for the clinical testing of an experimental drug to ensure that patient-reported outcomes are studied with adequate rigor. However, the agency also has a responsibility to prevent companies from making unsupported claims for drugs. Trial designs and endpoints for accelerated approvals in lung cancer. Dr. Fleming suggested that, if the literature suggests there is uncertainty about the reliability of DFS as a predictor of overall survival in the adjuvant setting, it might be more appropriate to use DFS as an endpoint for accelerated approval. Dr. Pazdur said that single-arm trials present analytical challenges, including the difficulty of analyzing toxicity, the inability to analyze time-to-event endpoints, and uncertainty about the minimal response rate that is acceptable. FDA is encouraging sponsors to conduct randomized trials that incorporate interim analysis and continue to confirm the existence of clinical benefit. However, this is a more expensive approach for sponsors. References 1. Johnson JR, Williams W, Pazdur R. End points and United States Food and Drug Administration approval of oncology drugs. J Clin Oncol 2003;21(7)(April):1404-1411. 2. Proc ASCO 2002: Raftopoulos, Bria, Gralla, Eid 3. Earle CC, Weeks JC (2003). The science of quality-of-life measurement in lung cancer. In Outcomes Assessment in Cancer, eds. J. Lipscomb, C.C. Gotay, C. Snyder. Cambridge: Cambridge University Press. 4. ASCO 2002 Abstract #1167 (measurement of QOL in IDEAL 2 randomized Phase 2 trial of gefitinib [Iressa] at two dose levels)
Date created: December 9, 2003 |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||