|
 |

|
|
 |
|||||
|
|||||||||
|
|
|
|
|
CDER Report to the Nation: 2004 Center for Drug Evaluation and
Research
CDER 2004 Report to the Nation: Print version
ContentsDirector's MessageIntroductionMissionHighlights and Initiatives
1 Drug Review
2 Drug Safety and Quality
3 International Activities
4 Communications
Director's MessageIntroductionWho we areThe Center for Drug Evaluation and Research is America’s consumer watchdog for medicine. We are part of one of the nation’s oldest consumer protection agencies-the Food and Drug Administration. The FDA is an agency of the federal government’s Department of Health and Human Services. We are the largest of FDA’s five centers, with about 1,800 employees. Approximately half of us are physicians or other kinds of scientists. Many of us have experience and education in such fields as computer science, legal affairs and regulatory matters. What we doOur best-known job is to evaluate new drugs for safety and effectiveness before they can be sold. Our evaluation, called a review, makes sure that the drugs we approve meet our tough standards for safety, effectiveness and quality. We also make sure that you and your doctor will have the information you need to use medicines wisely. Once drugs are on the market, we monitor them for problems. Reviewing drugs before marketing. A drug company seeking to sell a drug in the United States must first test it. We monitor clinical research to ensure that people who volunteer for studies are protected and that the quality and integrity of scientific data are maintained. The company then sends us the evidence from these tests to prove the drug is safe and effective for its intended use. We assemble a team of physicians, statisticians, chemists, pharmacologists and other scientists to review the company’s data and proposed use for the drug. If the drug is effective and we are convinced its health benefits outweigh its risks, we approve it for sale. We don’t actually test the drug when we review the company’s data. By setting clear standards for the evidence we need to approve a drug, we help medical researchers bring safe and effective new drugs to American consumers more rapidly. We also review drugs that you can buy over the counter without a prescription and generic versions of over-the-counter and prescription drugs. Watching for drug problems. Once a drug is approved for sale in the United States, our consumer protection mission continues. We monitor the use of marketed drugs for unexpected health risks. If new, unanticipated risks are detected after approval, we take steps to inform the public and change how a drug is used or even remove it from the market. We monitor changes in manufacturing to make sure they won’t adversely affect safety or efficacy. We evaluate reports about suspected problems from manufacturers, health care professionals and consumers. We try to make sure an adequate supply of needed drugs is always available to patients who depend on them Monitoring drug information and advertising. Accurate and complete information is vital to the safe use of drugs. We regulate information that accompanies or is displayed with an over-the-counter drug. In the past, drug companies promoted their products almost entirely to physicians. More frequently now, they are advertising directly to consumers. We oversee advertising of prescription drugs, whether to physicians or consumers. We pay particular attention to broadcast ads that can be seen by a great many consumers. We oversee advertising of prescription drugs, whether to physicians or consumers. The Federal Trade Commission regulates advertising of over-the-counter drugs. Advertisements for a drug must contain a truthful summary of information about its effectiveness, side effects and circumstances when its use should be avoided. Protecting drug quality. In addition to setting standards for safety and effectiveness testing, we also set standards for drug quality and manufacturing processes. We work closely with manufacturers to see where streamlining can cut red tape without compromising drug quality. As the pharmaceutical industry has become increasingly global, we are involved in international negotiations with other nations to harmonize standards for drug quality and the data needed to approve a new drug. This harmonization will go a long way toward reducing the number of redundant tests manufacturers do and help ensure drug quality for consumers at home and abroad. We also protect drug quality with rigorous manufacturing inspections to ensure compliance with current Good Manufacturing Practice requirements. Why we do itOur present and future mission remains constant: to ensure that drug products available to the public are safe and effective. Our yardstick for success will always be protecting and promoting the health of Americans. Getting consumer input. Protecting consumers means listening to them. We consult with the American public when making difficult decisions about the drugs that they use. We hold public meetings about once a week to get expert, patient and consumer input into our decisions. We also announce most of our policy and technical proposals in advance. This gives members of the public, academic experts, industry, trade associations, consumer groups and professional societies the opportunity to comment before we make a final decision. In addition, we take part in FDA-sponsored public meetings with consumer and patient groups, professional societies and pharmaceutical trade associations. These help us obtain enhanced public input into our planning and priority-setting practices. What is a drug?We regulate drugs used to treat, prevent or diagnose illnesses. However, drugs include more than just medicines. For example, fluoride toothpaste, antiperspirants, dandruff shampoos and sunscreens are all considered "drugs." You can buy some drugs in a store without a prescription, while others require a doctor's prescription. Some are available in less-expensive generic versions. Prescription drugsPrescription medicines must be administered under a doctor’s supervision or require a doctor’s authorization for purchase. There are several reasons for requiring a medicine be sold by prescription: n The disease or condition may be serious and require a doctor’s management. n The medicine itself may cause side effects that a doctor needs to monitor. n The same symptoms may be caused by different diseases that only a doctor can diagnose. n The different causes may require different medicines. n Some medicines can be dangerous when used to treat the wrong disease. Over-the-counter drugsYou can buy OTC drugs without a doctor’s prescription. You can successfully diagnose many common ailments and treat them yourself with readily available OTC products. These range from acne products to cold medications. As with prescription drugs, we closely regulate OTC drugs to ensure that they are safe, effective and properly labeled. Generic drugsA generic drug is a chemical copy of a brand-name drug. There are generic versions of both prescription and over-the-counter drugs. Generic drugs approved by the FDA have the same therapeutic effects as their brand-name counterparts. Scientific researchWe conduct and collaborate on focused laboratory research and testing. This maintains and strengthens the scientific base of our regulatory policy-making and decision-making. We focus on: n Drug quality, safety and performance. n Improved technologies. n New approaches to drug development and review. n Regulatory standards and consistency. MissionThe Center for Drug Evaluation and Research promotes and protects public health by assuring that safe and effective drugs are available to Americans. The Food and Drug Administration Modernization Act of 1997 affirmed the center's public health protection role, clarified the FDA's mission and called for the FDA to:
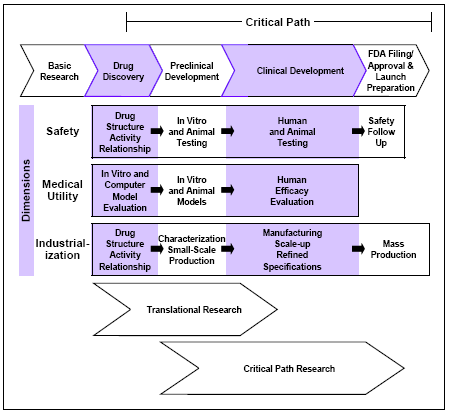
Highlights and InitiativesWe are pleased to present our ninth performance report. Our work in 2004 offered many Americans new or improved choices for protecting and maintaining their health or new ways to use existing products more safely. We worked hard at our mission of ensuring that Americans have safe and effective drugs and also developed these initiatives to bring the latest science and technology to bear on our mission: n Reforming our drug safety oversight. n Identifying steps to improve the science of drug development. n Improving manufacturing practices. n Protecting the homeland with improved medical countermeasures to be used in event of a terrorist attack or disaster. n Conducting targeted scientific research to improve our regulatory practices. We accomplished our work on these initiatives while maintaining our performance on our reviews of safety and efficacy and our oversight and surveillance of the safety of products sold to Americans. n Reviews. We approved 119 new medicines, including 36 truly new medicines that had not been marketed in any form before in this country. We approved 147 new or expanded uses for already approved medicines. We approved 380 generic versions of existing drugs. n User fee performance. We exceeded our goals for review performance. n Drug safety surveillance. We processed and evaluated more than 400,000 reports of adverse drug events, including more than 20,000 submitted directly from individuals. n Drug promotion and advertising. We issued more than 800 letters to help ensure manufacturers comply with regulations concerning drug promotion. n Bar codes on medicines. We promulgated a regulation that calls for bar codes on over-the-counter medicines commonly used in hospitals and most prescription medicines. n Public health advisories. We issued warnings on non-steroidal anti-inflammatory pain medicines and on antidepressant use in children, adolescents and adults. n Manufacturing. We implemented our initiative that encourages adoption of state-of-the-art manufacturing processes. Drug Safety InitiativeAmericans are rightly concerned about the safety of their drugs. Too many suffer from unexpected and unpreventable adverse events from the medicines they need. This results in human suffering and avoidable medical costs. Some have worried about “dangerous” drugs, while others have worried that an “overemphasis” on safety will delay developing new therapies. The most important concern for many Americans, however, has been the gap between the time a safety issue emerges and the time we know enough to make a regulatory decision. Our reforms of our drug safety efforts will: n Give patients, healthcare professionals and consumers quick and easy access to the most up-to-date and accurate information on medicines. n Make our drug review, approval and monitoring programs as transparent as possible. Drug safety has been and will continue to be a top priority for us. A recent internal audit showed that our professional staff spends about one-half its time addressing safety issues. Drug safety involves more than watching for problems once we approve a drug. Other important areas where the evaluation of drug safety takes place include: n Oversight of clinical trials. n Evaluation of safety and efficacy of new therapies and new or expanded uses for existing therapies. Because all drugs have risks, our evaluation must balance those risks against expected benefits. n Regulation of manufacturing, distribution and promotional activities. n Prevention of medication errors by evaluating proposed proprietary names, labeling and packaging. n Development of proactive risk management strategies both before and after approval. Oversight boardThe new, independent Drug Safety Oversight Board will oversee the management of drug safety issues and will provide emerging information to doctors and patients about the risks and benefits of medicines. It will: n Recommend information and updates for placement on the proposed Drug Watch Web page. n Resolve disagreements over approaches to drug safety issues. n Assess the need for MedGuides. n Oversee development and implementation of Centerwide drug safety policies. Oversight Board members don’t directly supervise approvalsThe board consists of FDA supervisors, members and medical experts from other medical agencies in the Department of Health and Human Services and other government departments such as the Department of Veterans Affairs. The board will consult with outside medical experts and representatives of patient and consumer groups. To avoid conflicts of interest, the board members have no direct supervision of approval decisions. Because board members are government employees, they will be able to freely discuss confidential and proprietary information. New communications channelsWe will share drug safety information sooner, more broadly and more conveniently through tailored drug safety information sheets for healthcare professionals and patients. We expect these new and direct communication channels will enhance knowledge and understanding of safety issues. Emerging or potential safety problems can be discussed even before we have reached conclusions that would prompt a regulatory action. The new communication channels include: n Drug Safety Web site. Consumers will find a variety of new information on specific drug products, including information sheets for patients and healthcare professionals (described below), the product’s regulatory history and its prescribing information. The site is http://www.fda.gov/cder/drugsafety.htm. n Proposed Drug Watch Web site. The Drug Safety Oversight Board will place “emerging” drug safety information on this site, such as possible serious side effects of particular drugs, before we have fully determined that the drug was responsible. This information will also include risks that might alter the benefit and risk analysis of a drug, affect patient selection, change monitoring decisions or could otherwise be avoided. n Healthcare professional information sheets. These will be one-page information sheets for all drugs on FDA’s Drug Watch and all drugs with Medication Guides. They will contain the most important new information for safe and effective product use, such as known and potential safety issues based on reports of adverse events, new information that may affect prescribing of the drug and the approved indications and benefits of the drug. n Patient information sheets. These one-page information sheets in a consumer friendly format will contain new safety information as well as basic information about how to use the drug for all products on Drug Watch. Ultimately, such sheets will be made available for every new drug that is approved. Institute of Medicine StudyWe have contracted with the IOM to study the effectiveness of the nation’s drug safety system. The study will have an emphasis on the post-market phase and assess what additional steps could be taken to learn more about the side effects of drugs as they are actually used. The IOM is the nation’s foremost body for science-based advice on matters of biomedical science, medicine and health. Differences of professional opinionIn most cases, free and open discussion of scientific issues among review teams and with supervisors, managers and external advisors, leads to an agreed course of action. Sometimes, however, consensus cannot be reached. We normally document these differences and their resolution in the official file of an action. An employee may sometimes feel that the normal process was inadequate and the decision made will have a significant negative impact on the public health. We have implemented a pilot process, coordinated by our ombudsman (click here), to provide for expeditious review and resolution of these differences by qualified staff not directly involved in the decisions. Critical pathCritical Path InitiativeFDA’s March 2004 report, Innovation or Stagnation?—Challenge and Opportunity on the Critical Path to New Medical Products, provides our analysis of the “pipeline problem.” There has been a slowdown—instead of the expected acceleration—in innovative medical therapies reaching patients. The medical product development path is becoming increasingly challenging, inefficient and costly. As a consequence, our mission to ensure the availability of safe and effective medical treatments for Americans that take advantage of the latest science is becoming compromised. In our view, the applied sciences for product development have failed to keep pace with the tremendous advances in the basic sciences. New science is not being used to guide the development process in the same way that it is accelerating the discovery process. To focus the attention of the public, academic researchers, funding agencies and industry, our report identifies: n The critical path for product development from design and discovery to commercial marketing. n The scientific and technical dimensions of the critical path. n The three types of research that support the critical path. Personalized medicineThe Critical Path recognizes the importance of “pharmacogenomics” and encourages its use in drug development. n Pharmacogenomics allows health care providers to identify differences in people’s drug response profiles and predict the best possible treatment options for them. Instead of a hit-or-miss approach to treating patients where it can take multiple attempts to find the right drug and the right dose, pharmacogenomics holds the promise that doctors will be able to analyze a patient’s genetic profile and prescribe the best available drug therapy and dose from the start. The field has experienced significant growth over the last few years. The sequencing of the human genome and the advent of new tools and technologies have already opened new possibilities in drug discovery and development. Personalized medicine for cancerGenomic tests are helping to identify cancers that have a good chance of responding to a particular medication or regimen. This has enabled the development of targeted therapies like trastuzumab for metastatic breast cancer, imatinib mesylate for chronic myeloid leukemia and cetuximab for metastatic colorectal cancer. Critical path dimensionsFrom the earliest phases of preclinical work to commercialization, developers must manage successfully in these three dimensions: n Assessing safety. Showing that a product is adequately safe for each stage of development. n Demonstrating medical utility. Showing a new product will actually benefit people. n Industrialization. Turning a laboratory concept into a consistent and well-characterized medical product that can be mass produced.
Research needed to improve development toolsTogether with academia, patient groups, industry and other government agencies, we need to embark on an aggressive, collaborative research effort to create a new generation of performance standards and predictive tools. We need tools that will provide better answers about the safety and effectiveness of investigational products, faster and with more certainty. We at FDA are uniquely suited to play a major role in this effort because of: n Our experience overseeing medical product development, assessment and manufacturing/marketing. n Our vast clinical and animal databases. n Our close interactions with all the major players in the critical path process. Public support for Critical PathWe have heard from patient groups, the drug and biotech industry, and industry groups. They concur with the scientific infrastructure problem and the focus on research, science-based standards and collaboration. Areas of major concern are: Clinical trialsn Statistical tools to support innovative trial design. n Disease modeling and trial simulation. n Standardization of trial administration. n Development of consistent electronic data collection, monitoring and reporting. Biomarkers and endpointsn Clarification of the process for validating surrogate endpoints. n Evidence needed to use biomarkers for other purposes, such as patient selection. n Standards for imaging as a biomarker. The way forwardThis initiative is not a fundamental departure for us, but rather builds on our proven best practices for developing industry guidance and expediting the availability of promising medical technologies. The next steps in this initiative include a series of workshops and meetings, to start development of a National Critical Path Opportunities list and to identify the key priorities. The full Critical Path report is available at http://www.fda.gov/oc/initiatives/criticalpath/whitepaper.html. You can view public comments to our proposal at http://www.fda.gov/ohrms/dockets/dockets/04n0181/04n0181.htm. Improving Manufacturing PracticesOur overhaul of our regulatory and quality control systems for pharmaceutical products encourages manufacturers to modernize their methods, equipment and facilities. Our goal is to help eliminate both production inefficiencies and undue risks for consumers. Our initiative implements improved policies that are making better use of our limited resources through more targeted and effective inspections. Collectively, our policies are known as “current good manufacturing practices” or cGMPs, and our last comprehensive revisions to them took place nearly a quarter of a century ago. Pharmaceutical cGMPs for the 21st Century is the umbrella name for this strategic initiative, and more information is available at http://www.fda.gov/cder/gmp/. Pharmaceutical cGMP initiative final report issuedIn 2004, we transitioned into an implementation phase and issued a final report on: n Our assessment of our regulations, current manufacturing practices as well the new tools in manufacturing science that will enable a progression to controls based on quality systems and risk management. n Specific steps we have taken and will take to develop and implement quality systems management and a risk-based product quality regulatory system. The report is available at http://www.fda.gov/cder/gmp/gmp2004/GMP_finalreport2004.htm#_Toc84065734. Process analytical technologies initiativeA key element of the cGMP initiative is our effort to encourage adoption of state-of-the-art quality control systems in manufacturing. This work is based on the premise that quality cannot be tested into products; it should be built into products by design. Process analytical technology is a system for design, analysis and control of manufacturing with the goal of ensuring final product quality. It does this through timely measurements—during processing—of critical quality and performance attributes of raw and in-process materials and processes. Effective use of the most current pharmaceutical science and engineering principles and knowledge—throughout the life cycle of a product—can improve the efficiencies of both the manufacturing and regulatory processes. Click here for more information. Emerging technologies in process validation recognizedWe revised a long-standing policy document regarding the validation of pharmaceutical manufacturing processes. New to this version is the recognition of the role of emerging advanced engineering principles and control technologies in ensuring batch quality. For drugs produced using these new principles and technologies, we provide for possible exceptions to the need for manufacturing multiple conformance batches prior to initial marketing. CounterterrorismThe first therapy for those exposed to a terrorism agent is often a drug. We have been taking an aggressive and proactive approach to our role in helping prepare the nation for terrorist events. These steps include: n Assuring the availability of medicines to treat victims of terrorist attacks. n Leveraging resources with other federal agencies to answer scientific questions concerning therapies to treat conditions caused chemical, biological or radioactive agents. n Preparing ourselves to continue operations during a crisis. n Protecting the nation’s drug supply from attack or deliberate contamination. Medical countermeasure approvalsn The infant atropine autoinjector (Pediatric AtroPen) provides an automatic injection of a potentially life-saving nerve agent antidote to children as young as 6 months. Doses and dosage forms of the AtroPen for adults and older children had been approved previously. n Pentetate calcium trisodium injection (Calcium DTPA) and pentetate zinc trisodium injection (Zinc DTPA) treat people who have become internally contaminated with certain radioactive isotopes (plutonium, americium or curium). The label includes information on pediatric dosing. These new molecular entities received priority approval and have orphan drug status. A second manufacturer received tentative approvals for these two drugs. n A second manufacturer of insoluble Prussian blue (Manoplex), to treat people internally contaminated with radioactive cesium-137 or thallium, received tentative approval in 2004. The first approval for insoluble Prussian blue was in 2003, and the product received orphan exclusivity. n We updated the ciprofloxacin (Cipro) label to include human information based on its use to prevent inhalational anthrax during the attacks in 2001. The label previously referenced only animal efficacy data for this indication. n Levofloxacin (Levaquin) is now approved to treat inhalational anthrax (post exposure prophylaxis) in adults. Levaquin is similar to ciprofloxacin, except it can be dosed once daily. n Fifteen new generic ciprofloxacin drug products were approved. Each will be indicated for prevention of inhalational anthrax post-exposure. n Penicillin G procaine injectable suspension (Wycillin) was approved to prevent the occurrence or progression of anthrax disease following exposure to Bacillus anthracis (including inhalational anthrax). Emergency use authorizationUnder the 2004 Project BioShield Act, we worked with the Centers for Disease Control and Prevention to identify potential medical countermeasures in the Strategic National Stockpile that we could authorize for emergency use for an unapproved indication. We also outlined the internal processes and procedures we need to handle an emergency use authorization. Emergency preparednessWe participated in four emergency response exercises. Threat agents were smallpox, anthrax, radiological contamination from a dirty bomb and cyanide. We also engaged in continuity of operations exercises, including an “at home” test to assure maintenance of vital operations and services in an emergency. Strategic National Stockpile regulatory, policy issues working groupWe participated in this internal FDA working group to address issues such as: n Compounding medical countermeasures during a mass casualty situation. n Labeling and dispensing medical countermeasures during a mass casualty. n FDA-shelf-life extension program and re-labeling. n Cities Readiness Initiative (mass prophylaxis dispensing). n Availability and vulnerability of products in the stockpile. n Risk assessment and enforcement discretion. n Proactive facilities inspections. n Patient access to life-saving therapies through investigational applications for countermeasures. Facilitating medical countermeasure developmentn Plague. The Centers for Disease Control and Prevention began enrolling patients in an FDA-funded clinical trial to assess the efficacy of the antibiotic gentamicin for endemic plague in two African countries where antimicrobial options for plague are extremely limited. We contributed to protocol design and the formation of a data safety monitoring board to oversee study safety concerns. We are continuing our collaboration with the Center for Devices and Radiological Health to evaluate the performance of a novel, rapid bedside plague diagnostic test kit under study conditions. n Pneumonic plague. We also continued our collaboration with the National Institute of Allergy and Infectious Diseases and the U.S. Army Medical Research Institute of Infectious Diseases to evaluate the safety and efficacy of five antibiotics (gentamicin, ciprofloxacin, levofloxacin, doxycycline and ceftriaxone) to treat pneumonic plague in a non-human primate model. Natural history studies, pharmacokinetic and toxicology studies to support efficacy studies and efficacy studies with high-dose and a humanized (lower) dose of gentamicin have been completed and analyzed. n Radiological and nuclear threats. We began another collaboration with the National Institute of Allergy and Infectious Diseases to identify promising new products for use against radiological and nuclear threats. We discuss scientific and regulatory issues with manufacturers of such products and inform them about possible funding sources, both for early development and for procurement by the federal government. Interagency collaborationsn Post-event surveillance planning. Along with the FDA’s other medical centers and the CDC, we developed a plan to identify processes for collecting adverse event and outcome data on medical products distributed in response to an emergency. n Project BioShield prioritization. We participated in many interagency working groups engaged in counter-terrorism efforts. These groups have contributed to gap analyses in medical countermeasures and have authored many of the requirements documents that will be used to prioritize products for development and eventual procurement under BioShield. Counterterrorism guidances published in 2004n Guidance for Federal Agencies and State and Local Governments: Potassium Iodide Tablets Shelf Life Extension. n Draft Guidance for Industry: Vaccinia Virus—Developing Drugs to Mitigate Complications from Smallpox Vaccination. Counterterrorism biotechnology researchWe have used congressionally mandated special funding to initiate research in several areas relevant to counterterrorism. Our scientists are studying: n Microarray technologies, which could assist in identifying infectious biowarfare agents. n Non-specific immune boosters, which could provide transient protection against such agents. n Monoclonal antibodies as neutralizers of biological toxins. n Various strategies to defend against anthrax. By establishing a core of scientists experienced in several areas of bioterrorism, these projects anticipate high-priority regulatory submissions likely to require rapid science-based evaluation. Internet resourcesWe provide links at http://www.fda.gov/cder/drugprepare/default.htm to the most current information on: n Drugs to prevent or treat disease caused by terrorism agents including drugs for use against anthrax, plague, radiation emergencies and chemical agents. n Drug development of counterterrorism products. n Vaccines. n Pediatric counterterrorism measures. n Prescribing and buying countermeasures. Scientific ResearchWe advance the scientific basis of regulatory practice by developing, evaluating or applying the best, most appropriate and contemporary scientific methods to regulatory testing paradigms. We provide scientific support for reviewer training, regulatory decision making and the development of regulatory policy. We focus on creating a tighter scientific linkage between non-clinical and clinical studies, enhancing methodology for assuring product quality, building databases for improved drug development and review and providing regulatory support through laboratory testing. Linking nonclinical and clinical studiesn Biomarkers for organ damage. We are identifying, evaluating and establishing relevant protein biomarkers in blood in both animal models and in humans. These will help detect the very earliest damage that can be caused by certain drugs to the heart, kidney, immune system and liver. n Biomarkers for inflammation. To enhance safety within broad segments of patient populations and enable safe development of new drug classes, we are working on the identification and elucidation of associated serum biomarkers and mechanisms responsible for the development of vascular inflammation in specific organ systems. n Evaluation of microarrays. We conduct targeted research on microarrays, a new technology that can identify thousands of genes or proteins rapidly and at the same time. We are evaluating how this technology could improve the interface between drug development and regulatory practice. n Medicinal plants, herbs. We established scientific research capabilities in the analyses of medicinal plant and herbal products. n Imaging drug targets. We continue to explore noninvasive imaging technology to extend our long-standing interest in the application of accurate dose-concentration-response principles by viewing drugs and their actions directly at the level of the drug target, rather than indirectly via plasma concentrations. n Better use of exposure-response data. We are developing a standardized approach for using exposure-response information to help evaluate the risks and benefits of drug therapies and recommending dose adjustments in special populations. n Pediatric pharmacokinetics. We are developing a pediatric population pharmacokinetics study design template to facilitate implementation of sparse sample strategies in pediatric drug development. Biotechnology researchOur new Office of Biotechnology Products was officially transferred to our center in 2003 from the Center for Biologics Evaluation and Research. The office consists of about 80 scientists and other staff who are responsible for evaluating therapeutic biotechnology product submissions as well as carrying out scientific research related to biologics regulatory issues. n Immune responses. We review many submissions aimed at inhibiting unwanted immune responses, such as autoimmune diseases or rejection of transplanted organs, or aimed at enhancing desired immune responses, such as those against infections or cancer. To facilitate review of such immunology-related submissions, we study the mechanisms by which immune cells are activated, suppressed or channeled from one kind of active response to another. n Metabolic pathways. We study the mechanisms by which various regulated products induce their intended effects, as well as unintended adverse effects. Our investigations also examine various normal and pathogenic pathways that are targeted by regulated agents. Our research enhances the ability of our scientist/regulators to evaluate risks and benefits of biotech products, to advise industry on difficult regulatory problems, such as potency assays, and to develop hands-on expertise in the modern technologies used by sponsors of biotech products. Informatics and computational safety analysisn Cancer toxicity predictive software. Our cooperative research and development agreements with several commercial software developers have resulted in the development and marketing of new computer software to predict the cancer-causing potential of chemicals based on their molecular structure. The software makes use of our extensive rodent carcinogenicity database without compromising proprietary information. n Safe starting dose models. We have successfully developed computer models to estimate the safe starting dose for clinical trials of drugs based on their molecular structure. The current method for estimating the starting dose is highly inexact and requires the use of multiple safety factors because it is based exclusively on an extrapolation from animal toxicity studies. We have begun studies to validate the new method. Scientific research in pregnancy and lactationClick here for studies to evaluate fetal safety from drug exposure or whether the dose of a drug should be adjusted during pregnancy or lactation. Laboratory supportOur efforts included: n Development of methods to evaluate quality attributes of drug products and raw materials by chemical imaging. These properties include polymorphic form, hydration state, stability and purity. n Rapid identification of counterfeit products using near-infrared spectroscopy and chemical imaging to discriminate drug products and raw materials. n Development of a methodology for the determining glove permeability to lindane shampoo and lotion, treatments for lice whose active ingredient is highly toxic. Pharmaceutical analysisWe collaborate with other organizations to ensure the availability of high quality standards and calibration materials. We collaborated with state pharmacy boards to evaluate Internet pharmaceuticals. We evaluated the quality of a select group of the most-often-ordered pharmaceutical products from foreign Internet suppliers.
Date created: Aug. 22, 2005 |