


FDA
Home Page | CDRH Home Page | Search
| CDRH
A-Z Index | Contact CDRH
![]()
|
| FDA
Home Page | CDRH Home Page | Search
| CDRH
A-Z Index | Contact CDRH
|
  |
| Information
for mammography facility personnel, inspectors, and consumers about the implementation of the Mammography Quality Standards Act of 1992 (MQSA) |
The Mammography Quality Standards Act Final RegulationsDocument #4: Final Guidance for Industry and FDADocument issued on May 23, 2001This document supersedes Draft Compliance Guidance: The Mammography Quality Standards Act Document #4 issued on  U.S. Department Of Health and Human Services Food and Drug Administration Center for Devices and Radiological Health Inspection Support Branch Division of Mammography Quality and Radiation Programs Office of Health and Industry Programs Preface Public Comment Comments and suggestions may be submitted at any time for Agency consideration to Dockets Management Branch, Division of Management Systems and Policy, Office of Human Resources and Management Services, Food and Drug Administration, 5630 Fishers Lane, Room 1061, (HFA-305), Rockville, MD, 20852. When submitting comments, please refer to the exact title of this guidance document. Comments may not be acted upon by the Agency until the document is next revised or updated. For questions regarding the use or interpretation of this guidance contact Charles Finder, M.D. at (301) 594-3332 or by email caf@cdrh.fda.gov. Additional Copies Additional copies are available from the Internet at:
TABLE OF CONTENTS
Background The Mammography Quality Standards Act Final Regulations Document #4
The Mammography Quality Standards Act (MQSA) was passed on October 27, 1992, to establish national quality standards for mammography. The MQSA required that to provide mammography services legally after October 1, 1994, all facilities, except facilities of the Department of Veterans Affairs, must be accredited by an approved accreditation body and certified by the Secretary of Health and Human Services (the Secretary). The authority to approve accreditation bodies and to certify facilities was delegated by the Secretary to the FDA. On October 28, 1997, the FDA published the MQSA final regulations in the Federal Register. The final regulations became effective April 28, 1999, and replaced the interim regulations (58 FR 67558 and 58 FR 67565). The FDA is using a variety of efforts to educate the public about the final regulations. These efforts include making presentations at key professional meetings and providing informational materials to the public. The currently available documents are included on our Internet home page (http://www.fda.gov/cdrh/mammography) containing all previously issued guidance, including the latest edition of "Preparing for the MQSA Inspection." This document is intended to provide guidance to mammography facilities and their personnel. It represents the Food and Drug Administration's (FDA) current thinking on the final regulations implementing the Mammography Quality Standards Act (MQSA) (Pub. L. 102-539). The FDA uses mandatory language, such as shall, must, and require, when referring to statutory or regulatory requirements. The FDA uses non-mandatory language, such as should, may, can, and recommend when referring to guidance. It is the responsibility of the facility to read, understand, and follow the final regulations. Under MQSA, a State may impose more stringent requirements beyond those specified by MQSA and its implementing regulations. A facility may want to check with the State or local authorities regarding their requirements. Question: Our group practice interprets mammograms sent to us by other facilities under a contractual arrangement. This is the only service that we provide in the mammography area. Does my group practice need an MQSA certificate to interpret mammograms? Answer: No. Partial providers (groups such as yours that provide only part of the services required for mammography) are certified as part of a "system" for producing, processing, and interpreting mammograms. The provider of some component of the system that performs mammography must take the lead in obtaining an MQSA certificate. FDA expects that, in most cases, the owner of an x-ray unit or units will apply for accreditation and receive an MQSA certificate. However, anyone interested in providing mammography services may apply for accreditation leading to an MQSA certificate. Under the facility accreditation process, partial providers, such as your group practice, are included in the required documentation provided to the accreditation body. The application for accreditation must show that all components of the system used to produce, process, and interpret mammograms meet MQSA requirements. These components do not have to be in the same location. If one or more of the facilities for which you (or wish to) interpret mammograms is applying for accreditation and certification (or reaccreditation and recertification), your responsibility will be to provide them with information documenting that the physicians in your group meet MQSA quality standards requirements for interpreting physicians. When the MQSA certificate is issued or renewed, all such partial providers are covered as providing services through that facility. Any partial provider providing services for more than one MQSA certified facility will be covered separately under each facility's MQSA certificate. Conversely, if one of the facilities for which you interpret mammograms is not certified, it would be unlawful for that facility to continue to produce mammograms and for your group to interpret mammograms produced by them. Your group practice could apply for accreditation and receive the MQSA certificate, as part of a system. In that case, your practice would be responsible for assuring not only that your practice meets MQSA quality standards for interpreting physicians, but also that the providers that produce and process the mammograms for your interpretation meet the quality standards that apply to them. Your practice would also be responsible for passing the review of clinical images from each facility that produces images for your interpretation, and for meeting the other requirements for accreditation. Finally, as an MQSA certificate holder, you would be responsible for paying an annual FDA MQSA inspection fee. In summary, it is the MQSA certificate holder who ultimately will be held responsible for assuring that all MQSA requirements are met. Personnel - General Question: What is the best way for personnel to document their mammography modality specific continuing education? Answer: The best way to document mammography modality specific continuing education is to have the education provider list the mammography modality credit hour "breakdown" on the CME/CEU certificate. Currently, most certificates do not provide such a "breakdown." FDA strongly encourages CME/CEU providers to provide this "breakdown" information on their certificates. Facilities are reminded that they are held responsible for maintaining documentation of their personnel's mammography modality specific continuing education and should have begun collecting this documentation as of April 28, 1999. For those CME/CEU certificates that do not list the mammography modality credit hour "breakdown," personnel may verify meeting this continuing education requirement by providing the CME/CEU certificate, a signed attestation form, and the course agenda or syllabus (see limited attestation policy) in the Policy Guidance Help System (PGHS). Note: Inspectors will not cite facilities for the mammography modality specific continuing education requirement before April 28, 2004. This will allow CME/CEU providers more time to provide mammographic modality specific documentation to their students. 21 CFR 900.12(a)(4) Question: We use the same locum tenens interpreting physician (or "temporary" radiologic technologist) on a recurring basis throughout the year. Do we need to handle the personnel records for this person differently from our "permanent" interpreting physicians (radiologic technologists)? Which records will be reviewed at the time of our next inspection? Answer: If the locum tenens interpreting physician (or "temporary" radiologic technologist) reads for (or performs mammograms for) the facility on a recurring basis throughout the year, the facility can treat this person as "permanent" for purposes of the annual inspection. In that event, the facility has to show that this person met all the requirements (including the continuing requirements, when appropriate) on the date of the inspection. If the facility considered this person "temporary" and not working at the facility at the time of the inspection, the facility has to show that this person met all the requirements (including the continuing requirements, when appropriate) on each of the multiple occasions this person started reading (or performing mammograms) at the facility. For example, if the person worked for a week at a time starting the 15th day of each month, the facility is responsible for documenting that the person was qualified on each of those 12 occasions. This is not as daunting a task as it first appears. The facility needs to document that the person was fully qualified the first time he/she started working at the facility. In addition, in each of the calendar quarters the person worked, the facility needs to document (before letting the person provide mammography services) that the person met the continuing requirements (when appropriate) and maintained a valid medical license (or valid State license or certification). Radiologic Technologist
Question: I have my ARRT (M) certificate. Will this certificate be sufficient documentation to show that I received training in breast anatomy and physiology, positioning and compression, quality assurance/quality control techniques and imaging of patients with breast implants as specifically spelled out in 900.12(a)(2)(ii)(A)? Answer: Yes. The ARRT (M) certificate will be accepted as documentation that the areas of training in breast anatomy and physiology, positioning and compression, quality assurance/quality control techniques and imaging of patients with breast implants have been covered. Keep in mind that by being awarded the ARRT(M) certificate, you have earned 24 contact hours toward the required total of 40 contact hours of training. Medical Physicist
Question: I have a masters (or higher) degree specifically in physics. Do I still need to document my number of semester hours in physics? Answer: If the degree is stated to be specifically in physics (or any of its sub-specialties), it will routinely be accepted during inspections as adequate documentation of the required number of semester hours in physics. Therefore, during routine inspections, you will not need to provide detailed transcripts to all the facilities where you provide physics services. If the degree was stated to be in the more general term, physical science, or in one of the three non-physics categories of physical science (chemistry, radiation science, or engineering), you will have to provide additional documentation demonstrating compliance with the appropriate requirement for semester hours in physics. 21 CFR 900.12(a)(3)(ii) Alternative Initial Qualifications.
Question: I have a bachelor's (or higher) degree specifically in physics. Do I still need to document my number of semester hours in physics? Answer: If the degree is stated to be specifically in physics (or any of its sub-specialties), it will routinely be accepted during inspections as adequate documentation of the required number of semester hours in physics. Therefore, during routine inspections, you will not need to provide detailed transcripts to all the facilities where you provide physics services. If the degree was stated to be in the more general term, physical science, or in one of the three non-physics categories of physical science (chemistry, radiation science, or engineering), you will have to provide additional documentation demonstrating compliance with the appropriate requirement for semester hours in physics. Equipment
Question: With machines such as the GE 500T and 600T, which do not have a separate mechanism for compression fine adjustment, can tapping the foot pedal for fine adjustment of compression force meet the year 2002 requirement? Answer: Yes. After receiving input from the National Mammography Quality Assurance Advisory Committee, comments from the public, and performing its own evaluation, FDA has determined that, with proper use, fine compression can be achieved with GE 500T and 600T units by tapping the foot pedal. While FDA recognizes that fine compression can be achieved using these mammography units, the specifics of the compression device require the technologist to pay additional attention during the application of compression. Where this causes clinical problems, facilities may want to consider modifying the compression device to allow for more consistent operator control. Facilities wishing to modify their units may contact their GE service representative for more information. Before a facility decides to modify the compression device, the facility should assure itself that the unit meets all the other new requirements (AEC performance, maximum compression force, focal spot condition and radiation output) that go into effect on October 28, 2002. 21 CFR 900.12(b)(10) Automatic exposure control.
Question: The regulations in 900.12(e)(5)(i) require that an x-ray unit pass an annual test for AEC performance over a range of 2 to 6 cm thick absorbers. If a unit is used clinically at combinations of kVp and filtration that include tissue thicknesses outside the 2 to 6 cm range, must it meet the AEC performance requirements at the thicknesses where it operates and must it be tested at those technique factors under the annual quality control requirements? Answer: No. The unit is not required to meet the AEC performance specification outside the 2 to 6 cm range and the physicist is not required to test the AEC performance requirements for thicknesses outside this range during the annual survey. However, we recommend that in addition to the required testing in the 2 to 6 cm range, the unit also be tested at all clinically used thicknesses outside this range and that the action limits specified in the regulations be applied to the extended test. If the unit cannot meet these action limits outside the 2-6 cm range, FDA recommends that a technique chart be developed showing appropriate techniques (kVp and density control settings) for the different breast thicknesses and compositions so that optical densities (OD) within +/- 0.30 OD (+/- 0.15 OD after October 28, 2002) of the average under phototimed conditions can be produced. Note:After October 28, 2002, the technique charts referred to in the preceding paragraph may be used only for thicknesses outside the 2-6 cm range. For use of technique charts within the 2-6 cm range. (See Question 1 under "AEC Performance Quality Control Test" in the PGHS regarding use of manual techniques when the AEC fails.) You should note that under the Equipment Evaluation outlined in 900.12(e)(10), an evaluation of the AEC under all clinically used configurations is required, not merely recommended (see next Q/A). This is because 900.12(e)(10) mandates conformance with all pertinent aspects of 900.12(b) and (e). Under 900.12(b)(10), the AEC is required to be "operable" under "configurations provided." In this context, "operable," as applied to the AEC means that it must meet the density and reproducibility requirements of (e)(5)(i) within the range of 2 to 6 cm. If designed to operate outside that range, the unit must meet the manufacturer's specifications over such additional ranges. Question: During an equipment evaluation, must AEC performance be tested for all equipment configurations used clinically by the facility or can it be limited to the contact configuration? What action limits apply? Answer: During an equipment evaluation, the AEC must be operable in all equipment configurations (e.g., grid, nongrid; magnification, nonmagnification; and various target-filter combinations) used clinically by the facility. Compliance with this requirement may be demonstrated by any of the following three methods:
Medical Records
21 CFR 900.12(c)(1)(v) In cases where no final assessment category can be assigned due to incomplete work-up, “Incomplete: Need additional imaging evaluation” shall be assigned as an assessment and reasons why no assessment can be made shall be stated by the interpreting physician; Question: What assessment category should be used for post lumpectomy patients, whose mammograms are otherwise negative? Answer: In the case described, the results could be categorized as either “negative” or “benign.” Where appropriate, qualifying statements such as “post surgical changes noted” may be added. The regulations require that each mammography report contain one of the six overall assessment categories. The decision as to which category to assign is left to the interpreting physician. Question: Is the use of an assessment code (e.g. 0, 1, 2, 3, 4, 5 or N, B, P, S, M, A) without the wording of the assessment category (e.g. Negative, Benign, etc.) sufficient to satisfy the reporting requirement? Answer: No. The wording of the assigned assessment category, chosen from one of the following; Negative, Benign, Probably Benign, Suspicious, Highly Suggestive of Malignancy, and Incomplete: Need additional imaging evaluation, must appear on the report. Question: We provide assessment categories for each breast (or for each lesion) of the examination. Do we still have to provide one overall assessment category for the entire examination? Answer: Yes. While individual assessments for each breast (or for each lesion), along with recommendations for their management, may be included in the report, one overall assessment category for the entire mammographic examination is required. It should be based on the assessment category of the more suspicious breast (or lesion). 21 CFR 900.12(c)(2)Communication of mammography results to the patient. Each facility shall send each patient a summary of the mammography report written in lay terms within 30 days of the mammographic examination. If assessments are ``Suspicious’’ or ``Highly suggestive of malignancy,’’ the facility shall make reasonable attempts to ensure that the results are communicated to the patient as soon as possible.
Question: What type of lay summary should be sent to a patient who has a normal mammogram, but the facility is aware that the patient has an abnormal physical exam? Answer: The facility has a great deal of flexibility in the format and substance of the lay summary. The lay summary must inform the patient of the results of the mammographic examination and follow-up actions, if needed. An acceptable lay summary could state that the mammogram was normal, but the patient should consult with the referring physician because of an abnormal physical exam. 21 CFR 900.12(c)(4)Record keeping. Each facility that performs mammograms:
Question: With the introduction of Full Field Digital Mammography, what constitutes a mammogram, the digital data or the hard copy film? Answer: There are two sections of the record keeping requirement that are affected by the introduction of digital mammography. The first deals with retention of the mammography films. For purposes of film retention, the facility must maintain, in retrievable form, either the digital data or hard copy films for the specified periods of time. For purposes of transferring films, the facility must be able to provide the medical institution, physician, health provider, patient or patient’s representative, with hard copy films of primary interpretation quality. Quality Control (QC) Tests - General Question: What are the minimum tests and/or reviews that the medical physicist must perform for a facility survey, survey of a mammography unit, equipment evaluation of a unit or processor that has been installed or disassembled and reassembled, and an equipment evaluation of a unit or processor that has undergone a major repair? Answer:
For more information, check additional guidance about the specific item being installed, reassembled or repaired. Question: While performing a facility survey, unit survey, or equipment evaluation, the medical physicist determines that the unit fails a required test(s) and needs to be adjusted, changed, or repaired. After the adjustment, change, or repair has been completed, does the test(s) have to be repeated and must the medical physicist perform the test(s)? Answer: After the adjustment, change, or repair has been completed, the test(s) have to be repeated and must show that the unit is within the appropriate action limit (verification test). For failures of the tests described in 900.12(e)(8)(ii)(A), 900.12(e)(6), 900.12(e)(7) and/or after adjustments, changes, or repairs that are classified as major, the verification test must be passed prior to using the item clinically. Note: verification testing performed after a major repair must be performed on-site by the medical physicist as required by 900.12(e)(10). For failures of the tests described in 900.12(e)(8)(ii)(B), the facility has a maximum of 30 days from the date of the initial test failure to correct the problem and pass the verification test. Facilities should have the verification test(s) performed (either by the medical physicist or by a qualified individual) as soon as possible after the adjustment, change, or repair is done, so as to allow time to repeat the adjustment, change, or repair if the equipment does not pass the verification test. Note: if the verification test is not passed within the 30 day time period, the item cannot be used clinically until such time as it is truly repaired and passes the test. For non-major equipment adjustments, changes, or repairs that are performed for any reason other than for failure of a required test, the facility must correct any problems found and perform verification tests to ensure that the equipment remains in compliance with the regulations. This should be accomplished as soon as reasonably possible. If the adjustment, change, or repair constitutes a “major repair (see Medical Physicist Involvement in Equipment Adjustments, Changes, or Repairs table later in this document),” the medical physicist must repeat the test(s) as part of the required follow-up equipment evaluation. If the adjustment, change, or repair does not constitute a “major repair,” the test(s) can be performed by any qualified person (e.g. radiologic technologist or service representative with appropriate training/experience). However, in this circumstance, FDA recommends that the medical physicist be consulted during this process. The following decision trees may be used to help in determining the timing and extent of physicist and facility involvement in response to test failures, equipment adjustments/changes/repairs and equipment evaluations.
*Note: In some cases, failures of the test(s) described in 900.12(e)(8)(ii)(B) and non-major adjustments, changes, or repairs may necessitate a verification test(s) described in 900.12(e)(8)(ii)(A). In those cases where an item fails its 900.12(e)(8)(ii)(A) verification test(s), the item cannot be used until such time as the verification test is passed. 
Question: Our facility has multiple mammographic units and uses several different screen-film combinations. What QC tests are we required to perform? Answer: The required QC tests are summarized in the following table. Table: QC Tests* Required for Facilities Using Multiple Units & Screen-Film Combinations
21 CFR 900.12(e)(1) Daily quality control tests. Film processors used to develop mammograms shall be adjusted and maintained to meet the technical development specifications for the mammography film in use. A processor performance test shall be performed on each day that clinical films are processed before any clinical films are processed that day. The test shall include an assessment of base plus fog density, mid-density, and density difference, using the mammography film used clinically at the facility.
Question: Must all facilities use 5 days to establish operating levels in a processor or can they use a longer period? Answer: The 5-day period is not a regulatory limit. Therefore, a facility may use a shorter or longer period if their situation calls for it. The number of days needed to establish the operating levels depends on the time it takes for the processor to reach chemical equilibrium or a "seasoned" status. In practice, this is usually achieved when the chemicals in the developer tank have been replaced or "turned over" 2-3 times. For a given film, this time is a function of the developer tank size, the replenishment rate, and the number of films processed (patient volume). Also, different film emulsion types have different replenishment rates per sheet of film, thus requiring different periods for the same processor to reach a seasoned status. Based on experience to date, we expect the majority of facilities to use up to a 5-day averaging period. However, if a facility uses a longer period, it should provide empirical data or recommendations from the film manufacturer explaining the reasons for the longer time period. Regardless of the length of time needed for establishing or re-establishing operating levels, the facility must document the MD, DD, and B+F values daily. Even though there are no regulatory action limits during this period, monitoring of processing conditions is still important as these values may identify problems with developer temperature, replenishment rates, and other variables. 21 CFR 900.12(e)(2) Weekly quality control tests. Facilities with screen-film systems shall perform an image quality evaluation test, using an FDA-approved phantom, at least weekly.
Question: Our facility has permanently glued the acrylic contrast disk to the phantom at the center of the cover plate. The regulation states that the OD be measured at the center of the phantom which would require our repositioning the disk (which may leave permanent artifacts) or replacement of the phantom cover. Is there another way to leave the disk in place and still fulfill the requirement? Answer: For the purpose of making the density measurements, FDA interprets measurements taken at the "center" of the phantom to be equivalent to measurements taken on the center line as described later in this paragraph. As an alternative to repositioning the disk or replacement of the phantom cover, you may choose to keep the disk in place (as is currently recommended by ACR). You may then measure the background density at a different location along the center line of the phantom in the direction parallel to the chest wall, as long as this location does not obscure any of the phantom objects. In addition, you must measure the background density at the same location each time the test is conducted. You should be aware that if you remove the disk, permanent defects may be produced in the cover plate. Such defects may produce "permanent artifacts" in the phantom image. As long as these "permanent artifacts" do not interfere with the scoring of the phantom (do not simulate masses, fibers or specks, and do not obscure the test objects), facilities may continue to use the phantom. The facility should not subtract these "permanent artifacts" from the phantom score. However, if these "permanent artifacts" interfere with the scoring of the phantom (simulate masses, fibers or specks, and/or obscure one or more of the test objects), the defective portion of the phantom must be replaced. The facility should also be aware that any artifacts on phantom images submitted for accreditation purposes may reduce the phantom image final score, which in some cases may result in a phantom image failure. 21 CFR 900.12(e)(4) Semiannual quality control tests. Facilities with screen-film systems shall perform the following quality control tests at least semiannually:
Question: We are a mobile facility with a van that does not have on-board processing. We have a film-changing room on the van where we load and unload cassettes with film during the day. When the van returns to our main office, we batch process the films. Do we have to perform a semiannual test for darkroom fog in this film-changing room? Answer: Yes. Since you use a room to load and unload cassettes with film, you must test this room for darkroom fog as well as the darkroom for your processor. Both rooms have the potential to fog films and degrade image quality. 21 CFR 900.12(e)(4)
Question: During clinical exams, how long must our mammography unit maintain the final compression force set by the radiologic technologist? Answer: The regulations do not specify a time frame in seconds or minutes. Instead, the minimum time frame is determined by the conditions at each facility. During clinical exams, the mammography unit must maintain the final compression force set by the radiologic technologist until the exposure is completed. If the radiologic technologist or interpreting physician notices that the compression force decreases significantly (such that the image quality is adversely affected) before the exposure is completed, the unit must be fixed, modified, or replaced. Question: When I perform the semiannual compression QC test, our unit's initial power drive provides an initial compression force of 37 pounds, however, it cannot maintain that force. The force decreases to less than 25 pounds in approximately 1 second. How long does the initial power drive have to maintain a force of at least 25 pounds? Answer: If the initial power drive is the sole means of providing compression for this mammographic unit, the unit must maintain a compression force of at least 25 pounds for the length of time it usually takes the radiologic technologist to complete an average exposure. If, during the semiannual compression QC test, the unit cannot maintain a force of at least 25 pounds for the specified timeframe, it fails the test and must be repaired, modified or replaced. If the unit passes this test, but still loses compression force during clinical examinations (see previous question), the unit must be repaired, modified or replaced. If the unit also has fine adjustment control (required on all units as of October 28, 2002), the initial power drive must maintain a compression force of at least 25 pounds for the length of time it usually takes the radiologic technologist to engage the fine adjustment control. The fine adjustment control must then maintain a compression force of at least 25 pounds for the length of time it usually takes the radiologic technologist to complete an average exposure. If, during the semiannual compression QC test, the unit cannot maintain a force of at least 25 pounds for the specified timeframe, it fails the test and must be repaired, modified or replaced. If the unit passes this test, but still loses compression force during clinical examinations (see previous question), the unit must be repaired, modified or replaced. QC Tests - Annual
Question: The regulations in 900.12(e)(5)(i) require that an x-ray unit pass an annual test for AEC performance over a range of 2 to 6 cm thick absorbers. If a unit is used clinically at combinations of kVp and filtration that include tissue thicknesses outside the 2 to 6 cm range, must it meet the AEC performance requirements at the thicknesses where it operates and must it be tested at those technique factors under the annual quality control requirements? Answer: No, the unit is not required to meet the AEC performance specification outside the 2 to 6 cm range and the physicist is not required to test the AEC performance requirements for thicknesses outside this range during the annual survey. However, we strongly recommend that in addition to the required testing in the 2 to 6 cm range, the unit also be tested at all clinically used thicknesses outside this range and that the action limits specified in the regulations be applied to the extended test. If the unit cannot meet these action limits outside the 2-6 cm range, FDA recommends that a technique chart be developed showing appropriate techniques (kVp and density control settings) for the different breast thicknesses and compositions so that optical densities within +/- 0.30 (+/- 0.15 after October 28, 2002) of the average under phototimed conditions can be produced. Note:After October 28, 2002, the technique charts referred to in the preceding paragraph may be used only for thicknesses outside the 2-6 cm range. For use of technique charts within the 2-6 cm range. (See Question 1 under "AEC Performance Quality Control Test" in the PGHS regarding use of manual techniques when the AEC fails.) You should note that under the Equipment Evaluation outlined in 900.12(e)(10), an evaluation of the AEC under all conditions of use is required, not merely recommended. This is because 900.12(e)(10) mandates conformance with all pertinent aspects of 900.12(b) and (e). Under 900.12(b)(10), the AEC is required to be “operable” under “configurations provided.” In this context, “operable,” as applied to the AEC means that it must meet the density and reproducibility requirements of (e)(5)(i) within the range of 2 to 6 cm. If designed to operate outside that range, the unit must meet the manufacturer’s specifications over such additional ranges. Question: During the annual physics survey, must AEC performance be tested for all equipment configurations used clinically by the facility or can it be limited to the contact configuration? What action limits apply? Answer: During the annual physics survey, AEC performance in the contact configuration must be tested. The medical physicist does not have to test the other clinically used equipment configurations during the annual physics survey, see guidance on determining AEC performance during equipment evaluations earlier in this document. In the contact configuration, the action limit requires maintenance of the film optical density (OD) over the 2-6 cm thickness range within +/- 0.30 OD (+/- 0.15 OD after October 28, 2002) of the mean.
Table 2
Question: Must the compression paddle be placed in the x-ray beam during half value layer (HVL) measurements? Answer: If the HVL measurement is being used to calculate patient dose, the compression paddle must be placed in the x-ray beam during the measurement. If the HVL measurement is being used for purposes other than patient dose, the position of the paddle is not specifically stated in the regulations. However, FDA recommends inclusion of the compression paddle in the x-ray beam because most manufacturers have designed their equipment to be tested in this configuration.
Question: Do all possible combinations of beam-limiting devices, image receptor sizes, target materials, and focal spots have to be tested during the annual x-ray field/light field/image receptor alignment quality control test? Answer: No. When performing the annual quality control test, the x-ray field/light field/image receptor alignment test must be performed only for those combinations of beam-limiting devices, image receptor sizes, target materials and focal spots used by the facility to produce full-field clinical images in the contact mode. For example, most facilities perform full-field clinical images using the large focal spot(s) in the contact mode. In this case, the alignment test has to be done only in the contact mode using the large focal spot(s). However, if, in addition to the large focal spot, the facility also routinely uses the small focal spot(s) to produce full-field clinical images in the contact mode, then the x-ray field/light field/image receptor alignment test must also be performed using the small focal spot(s). Facilities that only use the small focal spot to perform magnification or coned/spot images do not have to perform the alignment test in these configurations. Medical Physicist’s Annual Survey
21 CFR 900.12(e)(10) Mammography equipment evaluations. Additional evaluations of mammography units or image processors shall be conducted whenever a new unit or processor is installed, a unit or processor is disassembled and reassembled at the same or a new location, or major components of a mammography unit or processor equipment are changed or repaired. These evaluations shall be used to determine whether the new or changed equipment meets the requirements of applicable standards in paragraphs (b) and (e) of this section. All problems shall be corrected before the new or changed equipment is put into service for examinations or film processing. The mammography equipment evaluation shall be performed by a medical physicist or by an individual under the direct supervision of a medical physicist. Question: What constitutes an equipment evaluation (what tests must the medical physicist perform) for a processor that has undergone major repairs or is a new processor to the facility? Answer: At a minimum, the following tests must be done for a processor that has been replaced, undergone major repairs, or is a new processor to the facility: processor testing as described in 900.12(e)(1), phantom testing as described in 900.12(e)(2), and applicable portions of the system artifact evaluation as described in 900.12(e)(5)(ix). The medical physicist must also verify that the appropriate chemical solutions are being used, as described in 900.12(b)(13). If a change in clinical technique factors (for the standard breast) is involved that could significantly increase patient dose, a determination of dose as described in 900.12(e)(5)(vi) must be done. Question: Must the equipment evaluation report be sent to the facility within 30 days? Answer: The regulations do not specify when the equipment evaluation report must be sent to the facility. However, the facility cannot use the equipment until it has documentation (written preliminary or final equipment evaluation report) showing that the equipment passes all the appropriate tests. Question: The bucky assembly (cassette holder) is being replaced on our x-ray unit. Is it necessary that the medical physicist test and pass this prior to its use on patients? Answer: If the replacement includes the AEC detector, the medical physicist must evaluate the assembly prior to use on patients. If, however, the installation does not involve the replacement of the AEC detector(s) on the system, the exchange of bucky assemblies would not be considered a major repair and would therefore not require the medical physicist to evaluate the assembly prior to use on patients. There are tests that should be performed when the bucky is replaced (table artifacts, grid artifacts, and x-ray field/image receptor alignment). These particular tests require evaluation of images obtained with the new or modified equipment. In many cases, such images may be generated by facility personnel in consultation with the medical physicist but do not require the medical physicist to be physically present in the facility. The images may then be sent to the medical physicist for evaluation. If approved by the medical physicist, the facility may begin using the equipment. The medical physicist can then repeat the tests at the next annual survey. Question: Under what circumstances must our medical physicist actually visit our facility in order to successfully carry out his/her responsibility to oversee our equipment-related quality assurance practices? Answer: At a minimum, the medical physicist must be on-site to perform or to provide direct supervision for the performance of:
Determining when a medical physicist must be on-site in connection with adjustments, changes, or repair of equipment requires further discussion. Adjustments, changes, or repairs of equipment can occur as corrective actions for problems that caused the equipment to fail a quality control test, as the result of an unexpected equipment failure, or as a measure intended to prevent possible future inadequate equipment performance. All adjustments, changes, or repairs must include some form of verification testing to demonstrate that the affected equipment meets the applicable standards. As noted, in the case of major adjustments, changes, or repairs, the medical physicist is required to conduct a mammography equipment evaluation on-site to confirm that the applicable standards in 21 CFR 900.12(b) and (e) are met. FDA also recommends that the medical physicist have a role in some other changes or repairs through the provision of “medical physicist (MP) oversight.” By “MP oversight,” we mean that the medical physicist should be consulted as to whether an on-site visit is required or if other personnel can verify that the standards are met, with direction by telephone or printed material from the medical physicist if needed. Finally, FDA recognizes that there are adjustments, changes, or repairs for which verification (that the adjusted, changed, or repaired equipment meets standards) can be performed by other qualified personnel (e.g. radiologic technologist or service representative with appropriate training/experience) without involving the medical physicist. However, the facility can consult their medical physicist in these situations if they wish. The first column of the table, “Medical Physicist Involvement in Equipment Repairs,” lists the adjustments, changes, or repairs in which the regulations require a mammography equipment evaluation. It also includes a number of adjustments, changes, or repairs that do not require a mammography equipment evaluation. The second column indicates whether the item is considered by FDA to be a major adjustment, change, or repair. If so, it is stated in the third column that the “MP conducts evaluation in person.” For other adjustments, changes, or repairs, the third column indicates that FDA recommends either “MP oversight” or “MP involvement optional.” In cases where the recommendation is “MP oversight” or “MP involvement optional”, the verification may be accomplished by a qualified individual (as described in the previous paragraph) other than the medical physicist.
Table: Medical Physicist Involvement in Equipment Adjustments, Changes, or Repairs For any adjustment, change, or repair not listed in the table below, or if the facility is unsure as to the full extent of the adjustment, change, or repair, the facility should consult their medical physicist to determine the proper extent of his or her involvement in evaluating the item.
* Internal adjustments refer to equipment adjustments that typically cannot be made by the operator Question: For test failures, equipment evaluations and other adjustments, changes, or repairs, are there specific timeframes defining when the verification test(s) must be performed? Answer: For failures of the tests described in 900.12(e)(8)(ii)(A), 900.12(e)(6), 900.12(e)(7) and/or after adjustments, changes, or repairs that are classified as major, the verification test must be passed prior to using the item clinically. Note: verification testing performed after a major adjustment, change, or repair must be performed on-site by the medical physicist. For failures of the tests described in 900.12(e)(8)(ii)(B), the facility has a maximum of 30 days from the date of the initial test failure to correct the problem and pass the verification test. Facilities should have the verification test(s) performed (either by the medical physicist or by a qualified individual) as soon as possible after the adjustment, change, or repair is done, so as to allow time to repeat the adjustment, change, or repair if the equipment does not pass the verification test. Note: if the verification test is not passed within the 30 day time period, the item cannot be used clinically until such time as it is truly repaired and passes the test. For non-major equipment adjustments, changes, or repairs that are performed for any reason other than for failure of a required test, the facility must correct any problems found and perform verification tests to ensure that the equipment remains in compliance with the regulations. This should be accomplished as soon as reasonably possible. The following decision trees may be used to help in determining the timing and extent of physicist and facility involvement in response to test failures, equipment adjustments/changes/repairs and equipment evaluations. 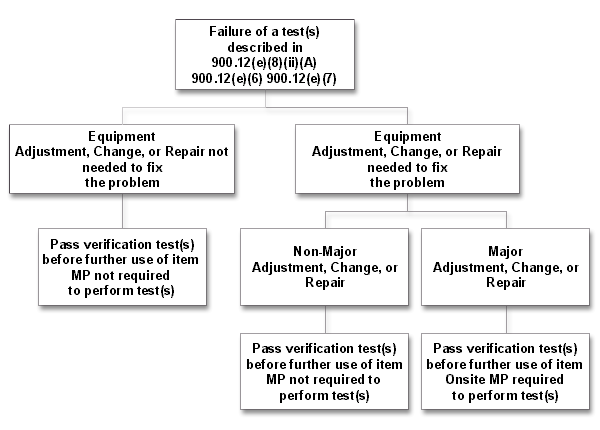
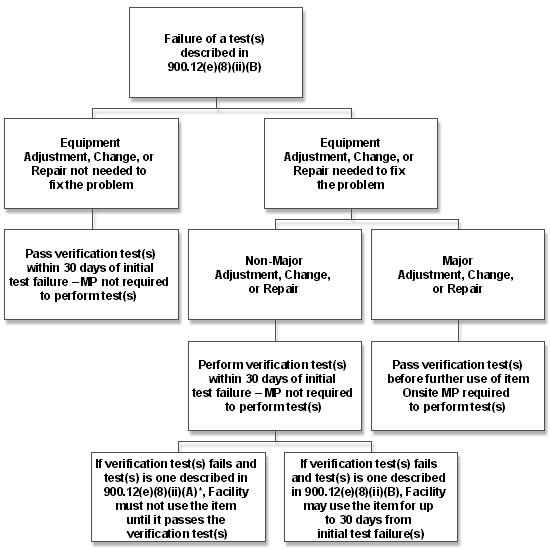

*Note: In some cases, failures of the test(s) described in 900.12(e)(8)(ii)(B) and non-major adjustments, changes, or repairs may necessitate a verification test(s) described in 900.12(e)(8)(ii)(A). In those cases where an item fails its 900.12(e)(8)(ii)(A) verification test(s), the item cannot be used until such time as the verification test is passed.
21 CFR 900.12(e)(13) Infection control. Facilities shall establish and comply with a system specifying procedures to be followed by the facility for cleaning and disinfecting mammography equipment after contact with blood or other potentially infectious materials. This system shall specify the methods for documenting facility compliance with the infection control procedures established and shall:
Question: We clean the mammography unit between each patient. Do we still have to document (through the use of logs or charts) cleaning after the equipment comes in contact with blood or other potentially infectious materials? Answer: The regulations require that equipment be cleaned and disinfected after contact with blood or other potentially infectious materials. Many facilities perform routine cleaning between all patients. However, in most cases, this routine cleaning is not sufficient to disinfect the equipment, either because different materials are required or the materials used are not left in contact with the equipment for a sufficient amount of time to achieve disinfection. Because of this, and the importance FDA places on this aspect of quality assurance, the facility is required to maintain a record of those instances when disinfection procedures were performed. Question: We use our mammography unit for interventional procedures as well as for routine mammography. Since interventional procedures are currently excluded from regulation, what should we be doing to document that proper infection control procedures have been performed? Answer: While interventional mammographic procedures are currently excluded from regulation, the regulations regarding the use of infection control procedures in patients undergoing non-interventional (“screening or diagnostic”) mammography are clear. The facility must document that the equipment was cleaned and disinfected after contact with blood or other potentially infectious materials, prior to use on these non-interventional mammography patients, regardless of how or when the equipment was contaminated. Question: We use our mammography unit solely for interventional procedures. Since interventional procedures are currently excluded from regulation, what are we required to do with respect to infection control procedures? Answer: Because interventional mammographic procedures are currently excluded from regulation, there are no requirements imposed on a facility that does not do “screening or diagnostic” mammography. However, because of the high probability of contamination with blood or other infectious materials during interventional procedures, FDA strongly recommends that such facilities document that they clean and disinfect their equipment after each of their patients. Mammography Medical Outcomes Audit Question: Are medical outcomes audit data collected during facility inspections used in a national database? Answer: No. While medical outcomes audit data are reviewed during the inspection to assure the facility is complying with the regulations, the data are not routinely collected by FDA. FDA does not maintain or supply information to such a national database. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Updated June 24, 2003
![]()
CDRH Home Page | CDRH A-Z Index | Contact CDRH | Accessibility | Disclaimer
FDA Home Page | Search FDA Site | FDA A-Z Index | Contact FDA | HHS Home Page
Center for Devices and Radiological Health / CDRH