


|
| FDA Home Page | CDRH Home Page | Search | A-Z Index |  |
|
For questions regarding this document, contact Thinh Nguyen (CDRH) at 240-276-4010 or by e-mail at thinh.nguyen@fda.hhs.gov. For questions regarding the application of this guidance to devices regulated by the Center for Biologics Evaluation and Research (CBER), contact Robert Yetter at (301) 827-0373 or by e-mail at Yetter@cber.fda.gov.
 |
|
U.S. Department of Health
and Human Services Food and Drug Administration Center for Devices and Radiological Health Center for Biologics Evaluation and Research |
Written comments and suggestions may be submitted at any time for Agency consideration to Dockets Management Branch, Division of Management Systems and Policy, Office of Human Resources and Management Services, Food and Drug Administration, 5630 Fishers Lane, Room 1061, (HFA-305), Rockville, MD, 20852. Alternatively, electronic comments may be submitted to http://www.fda.gov/dockets/ecomments. Please identify your comments with the docket number listed in the notice of availability that publishes in the Federal Register announcing the availability of this guidance document. Comments may not be acted upon by the Agency until the document is next revised or updated.
Additional copies are available from the Internet at: http://www.fda.gov/cdrh/mdufma/guidance/835.pdf or http://www.fda.gov/cber/mdufma/mdufma.htm, or by phone at (301) 827-2000 or (800) 835-4709, or to receive this document by fax, call the CDRH Facts-On-Demand system at 800-899-0381 or 301-827-0111 from a touch-tone telephone. Press 1 to enter the system. At the second voice prompt, press 1 to order a document. Enter the document number (835) followed by the pound sign (#). Follow the remaining voice prompts to complete your request.
III. Consultation with Stakeholders
VI. User Fee Considerations for Modular Review
VII. Industry Instructions for Submitting a Modular PMA
1. Determination that PMA Review is Appropriate for the Device
1. Submission of Each PMA Module
3. Time Frame for Reviewing a PMA Module
4. Reopening a Closed PMA Module
Attachment II Sample PMA Shell
Attachment III Frequently Asked Questions
Contains Nonbinding Recommendations
| This guidance represents the Food and Drug Administration's (FDA's) current thinking on this topic. It does not create or confer any rights for or on any person and does not operate to bind FDA or the public. You can use an alternative approach if the approach satisfies the requirements of the applicable statutes and regulations. If you want to discuss an alternative approach, contact the FDA staff responsible for implementing this guidance. If you cannot identify the appropriate FDA staff, call the appropriate number listed on the title page of this guidance. |
|
|
|||
| The purpose of this guidance
document is to provide industry and FDA staff with information regarding the premarket
approval application (PMA) modular review program and to outline the procedures for
submitting or reviewing a modular PMA. This guidance supersedes and replaces the documents entitled, "A Modular Approach to PMA Review," dated January 29, 1998, and "Guidance for the Medical Device Industry on PMA Shell Development and Modular Review," issued on November 6, 1998 (hereinafter referred to as the 1998 Guidances). FDA's guidance documents, including this guidance, do not establish legally enforceable responsibilities. Instead, guidances describe the Agency's current thinking on a topic and should be viewed only as recommendations, unless specific regulatory or statutory requirements are cited. The use of the word should in Agency guidances means that something is suggested or recommended, but not required. |
||||
|
|
|||
| In a traditional PMA, the applicant
submits all PMA data, as outlined in 21
CFR 814.20, at the same time, regardless of when testing is completed. FDA begins its
review only upon receipt of all the required information. In 1998, however, as part of
CDRH’s reengnineering effort, FDA issued the above mentioned guidances. In these
documents, FDA described a new policy whereby applicants could submit "Modular
PMAs." The goal of FDA’s 1998 Guidances was to increase the efficiency of the
PMA review process by allowing applicants to submit discrete sections (modules) of the PMA
to FDA for review soon after completing the testing and analysis. FDA intends the modular review approach to provide a mechanism by which applicants may submit preclinical data and manufacturing information for review while still collecting, compiling, and analyzing the clinical data. Therefore, a modular PMA is a compilation of sections or "modules" submitted at different times that together become a complete application. Additionally, the modular approach allows the applicant to potentially resolve any deficiencies noted by FDA earlier in the review process than would occur with a traditional PMA application. On October 26, 2003, the Medical Device User Fee and Modernization Act of 2002 (MDUMFA), Public Law 107-250 was enacted. Section 209 of MDUFMA amended the Federal Food, Drug and Cosmetic Act (the act) to codify the modular review approach.1 In recognition that the agency would need to issue updated guidance to reflect the new statutory provision, FDA requested comments on its modular PMA review program. On February 4, 2003, FDA published a Federal Register notice entitled, "Medical Device User Fee and Modernization Act of 2002, Establishment of a Public Docket (68 FR 5643)(hereinafter referred to as the MDUFMA Docket). In addition, FDA issued a guidance entitled, "Assessing User Fees: PMA Supplement Definitions, Modular PMA Fees, BLA and Efficacy Supplement Definitions, Bundling Multiple Devices in a Single Application, and Fees for Combination Products."2 In the guidance, FDA asked for comments on the various topics discussed in the document, including modular review of PMAs. Since 1998, FDA has gained considerable experience with modular PMA submissions. Based on FDA’s experience prior to the enactment of MDUFMA and comments submitted to the MDUFMA Docket, FDA is issuing this guidance to replace the 1998 Guidances. These changes include:
The agency expects these changes to: 1) help clarify which PMAs are appropriate for modular review, 2) simplify submission procedures, and 3) improve the efficiency of FDA’s review. Additionally, we have added guidance addressing user fee payment procedures for modular PMAs. |
||||
| _______________ 1 Section 515(c) of the act, as amended by section 209 of MDUFMA. 2 This guidance was issued on |
||||
|
||||
| We believe we should consider the
least burdensome approach in all areas of medical device regulation. This guidance
reflects our careful review of the relevant scientific and legal requirements and what we
believe is the least burdensome way for you to comply with those requirements. However, if
you believe that an alternative approach would be less burdensome, please contact us so we
can consider your point of view. You may send your written comments to the contact person
listed in the preface to this guidance or to the CDRH or CBER Ombudsman. Comprehensive
information on CDRH's Ombudsman, including ways to contact him, can be found on the
Internet at http://www.fda.gov/cdrh/resolvingdisputes/ombudsman.html and CBER’s Ombudsman can be reached at (301) 827-0379. |
||||
|
|
|||
| In developing this guidance, FDA has
considered comments on the PMA modular review program that were submitted to the MDUFMA
Docket. As mentioned above, FDA also issued the Assessing User Fees guidance and asked for
comments on modular review in that document.3 To date, FDA has received comments on modular PMA review submitted to the MDUFMA docket from one stakeholder. One comment requested that FDA expand the scope of modular review, now limited to original PMAs, to certain PMA supplements. In the agency’s experience, reviews of applications that are supplements to an already approved device do not ordinarily lend themselves to a modular format because the changes do not usually involve multiple modules. In addition, the agency currently does not have the resources that would be required to expand the program. Therefore, this guidance addresses procedures solely for the modular review of original PMAs. Another comment requested that FDA respond to incomplete modules in an abbreviated timeframe (i.e., 75 days rather than the 90 review that traditionally has been used by the agency). At this time, FDA believes that the current review timeframe represents a realistic projection of what the Food and Drug Administration's Center for Devices and Radiological Health and Center for Biologics Evaluation and Research can accomplish. However, as stated in the letter from the Secretary of Health and Human Services to Congress that accompanies the user fee legislation,4 FDA agreed to work with its stakeholders to develop appropriate performance goals for this program once it is fully implemented. FDA will also re-examine the modular review timeframe at that time. One comment requested that FDA assess user fees on modular PMAs only if the initial module was submitted on or after October 1, 2002. FDA agrees with this comment and will not assess user fees retroactively for modular PMAs first submitted before October 1, 2002. Finally, one comment requested FDA to develop a question and answer styled text explaining PMA modular review. We have done so and included it as an attachment to this guidance document. We continue to invite comments on this guidance. In addition, the agency intends to include the modular review program as a topic for discussion at future stakeholder meetings. |
||||
| _______________ 3 This guidance can be found on the CDRH website at: http://www.fda.gov/cdrh/mdufma/guidance/1201.html. 4 This letter can be found at: www.fda.gov/cdrh/mdufma/pgoals.html. |
||||
|
|
|||
| The PMA modular review program affords an alternative to the preparation, submission, and evaluation of traditional premarket approval applications (PMAs). Because FDA believes that PMA supplements would rarely be appropriate for modular review, the scope of this document is limited to applicants pursuing approval of original PMAs. | ||||
|
|
|||
| Modular PMA is a compilation
of sections or "modules" submitted at different times that together become a
complete PMA application. PMA Module is a discrete section of the PMA that can
be submitted and reviewed independently. A module is a set of elements, tests,
information, etc., that addresses a selected aspect of the device application, such as
manufacturing or animal testing. PMA Module Supplement is information submitted to a closed module for FDA review of a change or modification to the information provided in the original module. PMA Shell is an outline and description of the contents of all the modules that will comprise the PMA. |
||||
|
|
|||
| MDUFMA amends the act to provide FDA
new responsibilities, resources, and challenges. One significant provision of MDUFMA
permits FDA to collect user fees for certain premarket reviews (i.e., premarket approval
applications, premarket reports, supplements, premarket notifications, biologics license
applications, and efficacy supplements as discussed in more detail below) for those
applications received on or after October 1, 2002. On February 20, 2003, enabling
appropriations were enacted, thus allowing the agency to immediately begin to collect fees
for medical device applications. Please see http://www.fda.gov/oc/mdufma
for information on remitting user fees. As discussed in previous guidance on user fee payments for modular PMAs,5 the fee for a modular PMA is due upon submission of the first module. If FDA receives the first module prior to payment, we will place the file on hold until we receive payment and notify the applicant of this action by facsimile.6 FDA begins its review when the Office of Financial Management notifies CDRH or CBER that payment has been received. |
||||
| _______________ 5 Assessing User Fees: PMA Supplement Definitions, Modular PMA Fees, BLA and Efficacy Supplement Definitions, Bundling Multiple Devices in a Single Application, and Fees for Combination Products” can be found at: www.fda.gov/cdrh/mdufma/guidance/1201.html 6 Section 209 of MDUFMA authorizes FDA to put the application on hold until payment is received. |
||||
|
|
|||
Contact Review Group |
||||
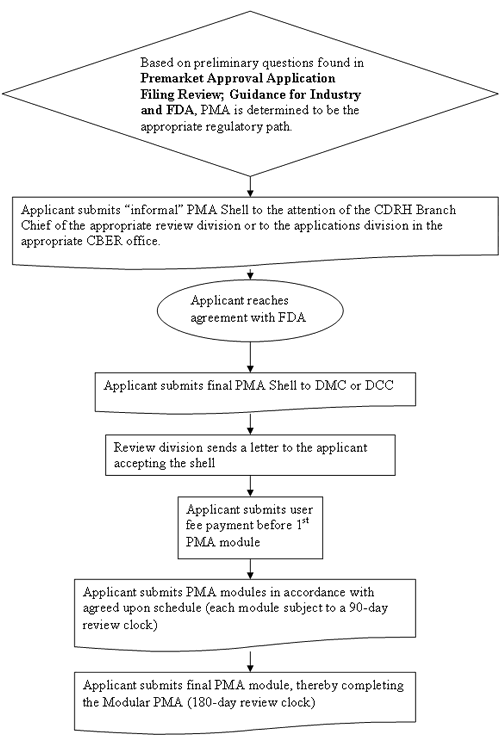
| Attachment I contains a flow chart of the steps ordinarily involved in modular PMA review. The first step in the modular PMA process should be contacting the CDRH Branch Chief within the appropriate review division or the applications division in the appropriate CBER Office to indicate your intention to submit a modular PMA.7 FDA believes early interaction with the appropriate branch/division and continued communication will optimize the opportunity for success of the modular PMA. | ||||
| ______________ 7 For assistance in identifying the appropriate review branch, the applicant may contact the Division of Small Manufacturers, International, and Consumer Assistance (DSMICA). Please see http://www.fda.gov/cdrh/devadvice/36f.html for contact options. For assistance in identifying the appropriate division in CBER, please contact the Office of Communication, Training and Manufacturers Assistance at 1-800-835-4709 or please see http://www.fda.gov/cber/inside/orgover.pdf. |
||||
|
|
|||
1. |
Determination that PMA Review is Appropriate for the Device |
|||
| Immediately upon receipt of an informal request for modular review, the Branch Chief assigns review staff (or other personnel designated by the division). That person answers a set of preliminary questions (please see Premarket Approval Application Filing Review; Guidance for Industry and FDA) aimed at ensuring that FDA utilizes the appropriate regulatory review path. Depending upon the answers to these preliminary questions, regulation as a class III device may be inappropriate. If the responses to the preliminary questions indicate that review of the modular proposal should not continue, staff notifies the applicant using the procedures identified in the guidance document referenced above. | ||||
|
|
|||
| FDA recommends that applicants use
the sample PMA shell (Attachment II Sample PMA Shell) as a model when designing PMA shell
proposals. In your PMA shell proposal, you should describe the contents of each module in
sufficient detail to provide FDA with a complete understanding of the modules you plan to
submit. The PMA shell proposal should address all elements required under 21
CFR 814.20 Application and the timing of each modular submission. If the clinical trial period will be lengthy or the product development time line is long, you should carefully consider your schedule for submitting PMA modules when developing your PMA shell. Premature submission of PMA modules (i.e., prior to finalizing your device design) could result in changes to the device that require you to submit additional data and FDA to re-evaluate closed PMA modules. FDA may refuse to accept a proposal for a PMA shell if the timing is inappropriate. For example, if you are within 6 months or less of submitting the final PMA module, FDA believes PMA modular review may not be appropriate because it may not allow sufficient time for FDA to complete its review of all the modules before receiving the final PMA module. In addition, with the exception of the manufacturing module, applicants should not plan to submit PMA modules in close succession. The schedule for submitting PMA modules should allow enough time for the review division to complete review of one module before the next module is received. Submitting PMA modules for concurrent review undermines the purpose of the modular review program because the agency cannot review multiple modules on an abbreviated cycle. If you intend to submit several modules at the same time or in close succession, FDA recommends that you submit a traditional PMA. The exception to this is the manufacturing module, which may be submitted at any time, regardless of when other modules were submitted. This is appropriate because the Office of Compliance, CDRH has primary responsibility for reviewing this data. Similarly, in CBER, the manufacturing section is reviewed by the Office of Biologics Quality. Therefore, FDA can efficiently concurrently review the manufacturing module, while another module is being reviewed. The manufacturing information should be submitted as a separate module to allow for ease of distribution to and review by the appropriate office. Please notify FDA, in the form of an amendment to your shell, when you anticipate that you are within 90 days of submitting your final PMA module, so that FDA can schedule a pre-approval Quality System Regulation inspection. |
||||
|
|
|||
| After you contact the appropriate Branch Chief, and it is agreed that PMA is the appropriate regulatory path, you should fax or email a draft of the proposed PMA shell to the branch chief to allow for review and comment on the proposal. The Branch Chief assigns a staff member to review the proposed shell. This individual is responsible for communicating to you any FDA recommended changes and reaching verbal agreement with you on the shell.8 | ||||
| ____________ 8 If agreement cannot be reached with the review staff assigned to review the shell within 30-days, please contact branch/division management. |
||||
|
|
|||
| After you reach agreement with FDA
on the proposed PMA shell, you should submit the finalized shell to the following address,
as applicable: PMA Document Mail Center (HFZ-401) Or to CBER at: Food and Drug Administration In order to facilitate the log-in process, you should prominently identify the name of the review staff member with whom you have been interacting in your cover letter. After the FDA receives the PMA shell and assigns a unique document control number (shell number), the review staff will confirm that the PMA shell is consistent with the agreed upon proposal and issue an acceptance letter (usually within 2 weeks). You may then submit PMA modules for review, according to the agreed upon schedule. |
||||
|
|
|||
| If you need to make changes to the shell after it has been accepted (e.g., device design modifications are made that require additional or different testing or the number of necessary modules increases or decreases), you should submit an amendment to the shell with a description of the changes and an explanation of the need for changes. After reviewing these changes, FDA may wish to discuss them with you. Once verbal agreement has been reached, you should submit the revised shell to the appropriate address listed above. | ||||
|
|
|||
1 |
Submission of Each PMA Module |
|||
When submitting a module, you should
clearly identify the submission as a module and reference the previously assigned shell
number (and module number) in the cover letter. As outlined in Attachment
II Sample PMA Shell, you should include the following in every module:
Duplication of some information between modules is necessary to allow the review staff assigned to that PMA module to review it in an efficient manner. |
||||
|
|
|||
| The full benefit of PMA modular review to both FDA and the applicant cannot be realized if PMA module submissions are routinely incomplete. Therefore, you should complete all testing to support the specific PMA module before submitting the module to FDA for review. FDA may reject an incomplete module by issuing a letter to the applicant stating that the review may not proceed until we receive the missing information. FDA intends to review each module within a 90-day time period, beginning upon submission of an amendment containing the balance of the missing information. | ||||
|
|
|||
| FDA’s objective is to complete
the review of each module and issue a deficiency letter or an acceptance letter within 90
days of receipt of the module. Deficiency letters notify the applicant of outstanding
issues that need clarification or additional information. Acceptance letters notify the
applicant that all issues have been resolved. FDA intends to review any amendments responding to a deficiency letter under a 90-day clock. Submitting any unsolicited major amendment at any time during the review of the PMA module resets the 90-day clock. Major amendments are defined in 21 CFR 814.37(c)(1). |
||||
|
|
|||
| If you modify your device after FDA deems a PMA module acceptable, you should contact the appropriate review division staff to discuss the modification and any testing needed to support it. You should submit data generated to support the modification separately to each of the affected modules with a revised executive summary. FDA logs-in this type of submission as a supplement to the PMA module. As described above, FDA intends to issue a deficiency or acceptance letter within 90 days of receipt of the supplement. | ||||
|
|
|||
| An applicant’s submission of the
final PMA module (i.e., final clinical data, proposed labeling, and summary of safety and
effectiveness), plus the incorporation by reference of previously submitted modules, will
complete the modular PMA. You should clearly identify the final PMA module submission as
the "COMPLETED MODULAR PMA" in the cover letter. You should
specifically reference (by shell and module numbers) the modules that have been accepted
by the FDA and also identify any modules with outstanding deficiencies at the time of PMA
submission. In order for the PMA to be complete, responses to all outstanding deficiencies
related to previously submitted modules should accompany the final PMA module. Upon receipt of this final PMA module, FDA assigns a PMA number to the completed application. |
||||
|
|
|||
| Upon receipt of the final module, FDA makes its filing decision. This decision is based on whether the last module includes all the information necessary to complete the PMA as required by 21 CFR 814.20. The PMA Filing guidance9 describes the kind of information required under this regulation that needs to be submitted before FDA can file a PMA. FDA plans to use the information described in this guidance in making filing determinations and, as for a traditional PMA, generally issues a filing/non-filing letter within 45 days of receipt of the last module. If FDA decides to file the PMA, the filing date is the date that the application became complete, typically the receipt date of the last module. The 180-day "PMA clock" under 21 CFR 814, also begins on that date. If the agency decides not to file the PMA, FDA will issue a non-filing letter to the PMA applicant. | ||||
| ____________ 9 See “Premarket Approval Application Filing Review” at www.fda.gov/cdrh/ode/guidance/297.html |
||||
Company/Device Name |
||
| Module # | Contents | Projected Date of Submission |
| Module 1 | Table of Contents for Module 1 Executive Summary* Device Description and Principles of Operation Declaration of Conformance to Standards for Module 1 Bibliography/References for Module 1 Non-clinical Laboratory Studies, for example:
|
|
| Module 2 | Table of Contents for Module 2 Executive Summary* Device Description and Principles of Operation Declaration of Conformance to Standards for Module 2 Bibliography/References for Module 2 Non-clinical Laboratory Studies, for example:
|
|
| Module 3 | Table of Contents for Module 3 Executive Summary* Device Description and Principles of Operation Manufacturing Information
|
|
Final PMA Module |
Table of Contents
for entire PMA, including all modules
SSED (i.e., compilation of executive summaries) Clinical Data (including Protocols, Results and Analyses) Financial Disclosure Information Proposed Labeling:
Post-marketing Plan (e.g., proposed long-term follow up studies, if appropriate) Bibliography/References for the Final PMA Module | |
________________________
* Executive Summary should contain a summary of the testing and results provided in the module.
Attachment III Frequently Asked Questions |
1. What is the "sample Modular PMA Shell"?An example of a sample Modular PMA shell is provided in Attachment II of this guidance document. FDA recommends that you follow this model whenever possible. Ideally, there should be no more than 3-4 modules. However, we understand that there may be instances when the recommended model is not suitable and should be modified. The review staff will work with you to establish a shell that is acceptable to both parties. |
|
|
| __________________ 10 Please discuss with the appropriate regulatory project management branch in CBER. |
|
|
|
| _________________ 11 [1] “Assessing User Fees: PMA Supplement Definitions, Modular PMA Fees, BLA and Efficacy Supplement Definitions, Bundling Multiple Devices in a Single Application, and Fees for Combination Products” can be found at: www.fda.gov/cdrh/mdufma/guidance/1201.html 12 See section 515(c)(3)(A) of the act. |
Updated November 3, 2003
****Accessible Text for Attachment I Above:
![]()
CDRH Home Page | CDRH A-Z Index | Contact CDRH | Accessibility | Disclaimer
FDA Home Page | Search FDA Site | FDA A-Z Index | Contact FDA | HHS Home Page
Center for Devices and Radiological Health / CDRH
