|
 |
 |
|
 |
FDA Home Page | Search FDA Site | FDA A-Z Index | Contact FDA

Food and Drug Administration
Department of Health and Human Services
for the


Printable version (556 KB PDF)
MDUFMA Commitments: Goals and Approaches
FY 2007 Activities and Accomplishments
MDUFMA Performance At-A-Glance for FY 2003 through FY 2007
Reauthorization of MDUFA for FY 2008 through FY 2012
Report on FY 2007 MDUFMA Performance
PMAs, Panel-Track PMA Supplements, and Premarket Reports
Expedited PMAs
180-day PMA Supplements
510(k) Premarket Notifications
BLAs
BLA Supplements
Resubmitted BLAs and BLA Efficacy Supplements
Additional MDUFMA Performance Commitments
Appendices
Appendix A: November 14, 2002, Commitment Letter from The Department of Health and Human Services (HHS) Secretary to Congress
Appendix B: Measuring Performance Under MDUFMA
Appendix C: Summary of MDUFMA’s Quantitative Goals
I am pleased to report that the Food and Drug Administration (FDA) continues to succeed in improving the process for the review of medical device applications and meeting the performance goals established under the Medical Device User Fee and Modernization Act of 2002 (MDUFMA).
MDUFMA requires close collaboration with stakeholders and increased communication with applicants. FDA is working to clarify its regulatory requirements and make its decisions more transparent through new guidance, educational materials, and meetings. We continually seek to reduce the burdens associated with device reviews and to enhance the efficiency and flexibility of our review processes. These efforts help applicants improve the quality of their submissions, and help FDA provide timelier, better-focused reviews. Our ultimate objective — an objective we share with industry — is to make important new medical devices available to patients and healthcare providers earlier, while continuing to ensure the quality, safety, and effectiveness of those devices.
I am proud of the significant progress FDA has made in meeting the challenges and responsibilities provided by MDUFMA. I believe the results we have achieved, and the long-term objectives we continue to pursue, clearly demonstrate the value of this important legislation to FDA, to the medical device industry, and, particularly, to patients and healthcare professionals. I am pleased that the medical device industry and Congress share this view, as evidenced by the enactment of the Medical Device User Fee Amendments of 2007 (MDUFA) reauthorizing medical device user fees for fiscal year (FY) 2008 through FY 2012. FDA is looking forward to 5 more years of cooperative efforts to make important new medical devices available to healthcare professionals and patients.
Andrew C. von Eschenbach, M.D.
Commissioner of Food and Drugs
MDUFMA amended the Federal Food, Drug, and Cosmetic Act (FD&C Act) authorizing FDA to collect user fees from manufacturers who submit certain applications to market medical devices. In parallel with this authority, MDUFMA required FDA to pursue a comprehensive set of review performance goals and commitments from FY 2003 through FY 2007 to improve the timeliness and predictability of medical device reviews and to improve communications between FDA and industry.
FDA has made good progress in meeting MDUFMA’s objectives and performance goals. FDA has worked with stakeholders to improve communication and understanding of MDUFMA requirements and to ensure that implementation accomplishes MDUFMA objectives. The performance gains and improved predictability in review processes achieved under MDUFMA are leading to significant benefits to industry, healthcare professionals, and patients.
FY 2007 Activities
FDA continued to focus on consulting with its stakeholders, developing guidance documents, and designing and building the new review processes required to meet MDUFMA’s challenging performance goals. Among the key activities during FY 2007 were:
FDA’s overall performance to date for the FY 2003 through FY 2007 receipt cohorts indicates FDA is meeting or exceeding most MDUFMA performance goals. Of the 77 quantifiable MDUFMA performance goals that were in effect for the FY 2003 through FY 2007 cohorts, FDA’s performance to date includes meeting or exceeding 47 goals and not meeting 14 goals (only one of these involves a decision goal and most of the other unmet goals involve measures based on a small number of applications or actions). The remaining 16 goals did not have actions completed as of September 30, 2007. As additional FDA decisions are made, particularly for more recent cohorts, FDA expects these preliminary results to change.
“…prompt approval and clearance of safe and effective devices is critical to the improvement of the public health so that patients may enjoy the benefits of devices to diagnose, treat, and prevent disease…”
-- Section 101(1) of the Medical Device User Fee and Modernization Act of 2002.
On October 26, 2002, MDUFMA was signed into law. MDUFMA amended the FD&C Act authorizing FDA to collect fees from companies who submit certain applications for marketing of medical devices. In return, MDUFMA requires FDA to pursue a comprehensive set of device review performance goals that are intended to significantly improve the timeliness and predictability of FDA’s review of new devices.1 These performance goals were developed collaboratively and are defined in the HHS Secretary’s November 14, 2002, letter to Congress.2 Information about MDUFMA, including the text of the amendments and the performance goals and procedures, can be found on FDA’s Web site at: http://www.fda.gov/oc/mdufma.
On April 1, 2004, MDUFMA was amended and expanded by the Medical Device Technical Corrections Act (MDTCA), Public Law (P.L.) 108-214. MDTCA amended MDUFMA to clarify Congress’ intent and to improve and expand upon some features of MDUFMA. These changes did not affect the performance goals FDA was pursuing under MDUFMA. On August 1, 2005, the Medical Device User Fee Stabilization Act of 2005 (the “Stabilization Act”), P.L. 109-43, amended provisions of the FD&C Act relating to medical device user fees and device labeling.
On September 27, 2007, the President signed FDAAA, which included MDUFA. MDUFA reauthorizes medical device user fees and identifies new performance goals that go into effect from FY 2008 through FY 2012.
MDUFMA requires the HHS Secretary to submit two annual reports to Congress for each fiscal year fees that are collected: 1) a performance report due within 60 days of the end of the fiscal year, and 2) a financial report due within 120 days of the end of the fiscal year. This report is FDA’s fifth annual performance report on its progress in achieving MDUFMA performance goals and additional commitments, and covers actions through FY 2007.
The goal of MDUFMA is to better serve the public health by providing additional funds to FDA for “the process for the review of devices and the assurance of device safety and effectiveness so that statutorily mandated deadlines may be met.” The user fees provided by MDUFMA, and the additional appropriations that go with the new law, will provide the following significant benefits:
In addition to authorizing FDA to collect user fees for medical device applications, MDUFMA established review performance goals for FDA. These goals are intended to achieve progressive, year-by-year, improvements in review processes for medical devices. Few objectively-measurable goals were applied during FY 2003 and FY 2004, allowing FDA time to hire staff, build infrastructure, provide guidance to industry, and take other actions before it would be possible for FDA to begin to make substantial progress in improving overall review performance. Consequently, most substantive review performance goals went into effect in FY 2005. More goals went into effect in FY 2006 and again in FY 2007, with the goals becoming more demanding each year (see Appendix C for an overview of MDUFMA’s objectively-measurable performance goals).
The majority of devices associated with MDUFMA are reviewed by the Center for Devices and Radiological Health (CDRH). However, a number of devices that are critical to ensuring the safety, purity, and potency of biologic products, including assuring the safety of our Nation’s supply of blood and human tissue products, are reviewed by the Center for Biologics Evaluation and Research (CBER). Additionally, CBER regulates diagnostic tests for retroviruses, including human immunodeficiency virus , as well as devices used in cell and gene therapies. An Intercenter Agreement between CBER and CDRH discusses the types of devices regulated by CBER and is available at: http://www.fda.gov/oc/ombudsman/bio-dev.htm.
This report is concerned primarily with the performance goals that are an integral part of MDUFMA. FDA has prepared a summary of MDUFMA, including information on topics not covered by this report; see http://www.fda.gov/cdrh/mdufma/mdufmasummary.pdf. FDA also prepares an annual financial report that provides information on review fee revenues and expenses and compliance with MDUFMA requirements concerning the collection and use of those fees; the current and past reports are available at: http://www.fda.gov/oc/mdufma.
MDUFMA has three particularly significant provisions related to FDA performance:
MDUFMA made several other significant changes, including:
FDA made steady progress in implementing MDUFMA in FY 2007. FDA continued to focus on consulting with its stakeholders, developing guidance documents, and building the new review processes required to meet MDUFMA’s progressively challenging performance goals. Among the key activities and accomplishments during FY 2007 were:
A preliminary performance assessment of FY 2003 through FY 2007 submissions subject to MDUFMA goals and acted on as of September 30, 2007, indicates that FDA is meeting or exceeding most MDUFMA performance goals (see tables below).3
With the reauthorization of the medical device user fees for FY 2008 through FY 2012, MDUFA commits FDA to meeting new premarket review performance goals. These performance goals were developed with input from industry and are a key part of the negotiated package of user fees and other changes made by the 2007 MDUFA amendments. The new performance goals focus strongly on FDA decisions, because FDA decisions are so strongly linked to the final approval or clearance of new devices. As a key step in transitioning to the new performance goals, FDA will continue to work to meet MDUFMA’s decision goals for submissions received in FY 2007 and prior years. FDA will no longer apply, track, or report on MDUFMA’s first-action and subsequent-action cycle goals. During FY 2008, FDA will focus on implementing and reporting on the new performance goal requirements under MDUFA.
This section presents FDA's preliminary performance on MDUFMA performance goals and commitments for FY 2007. Additionally, performance data for FY 2003 through FY 2006 presented in previous MDUFMA performance reports has been updated to include actions FDA completed during FY 2007. The following information refers to FDA performance presented in this section.
Performance goals. MDUFMA requires that FDA meet the following types of performance goals:
Additional commitments. In addition to the performance goals, MDUFMA holds FDA to several commitments related to the medical device review process. These include programs and activities related to the application of user fee revenues, guidance development for the modular PMA review program, and examination of FDA’s bundling policy. Additional information on these commitments is presented in section I of FDA’s Commitment Letter in Appendix A.
Measuring performance. Progress on MDUFMA performance goals and commitments is measured in different ways, based on the type of goal or commitment. The following types of measures are used to capture FDA’s progress on meeting MDUFMA performance goals and commitments:
A more detailed description of performance measures is presented in Appendix B.
The table below summarizes the annual review time goals for PMAs, panel-track PMA supplements, and premarket reports. One cycle goal (30 days) became effective in FY 2003. Four additional cycle goals became effective in FY 2005 with the performance levels increasing incrementally through FY 2007. The 320-day decision goal became effective in FY 2006 with the performance level increasing to 90 percent in FY 2007. The 180-day decision goal became effective in FY 2007.
| Goals | Review Time Goal (Days) |
Performance Level | ||||||
|---|---|---|---|---|---|---|---|---|
| FY 03 | FY 04 | FY 05 | FY 06 | FY 07 |
||||
| Decision | Make an "FDA decision" | 320 | No Goal | 80% | 90% | |||
| 180 | No Goal | 90% | ||||||
| Cycle | Issue a "major deficiency" letter as the first action | 150 | No Goal | 75% | 80% | 90% | ||
| Issue all other first actions | 180 | No Goal | 75% | 80% | 90% | |||
| Issue a "major deficiency" letter as the second or later action | 120 | No Goal | 75% | 80% | 90% | |||
| Act on an amendment containing a complete response to a "major deficiency" or "not approvable" letter | 180 | No Goal | 75% | 80% | 90% | |||
| Act on an amendment containing a complete response to an "approvable" letter | 30 | 90% | ||||||
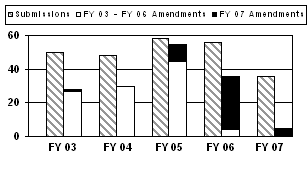
The number of PMA and panel-track PMA supplements submitted decreased to a 5-year low in FY 2007.4 Additionally, 48 amendments were received in FY 2007 with most (32) applying to the FY 2006 cohort (see corresponding graph and table).
PMAs, Panel-track PMA Supplements, and Premarket Reports
| Type | FY 03 | FY 04 | FY 05 | FY 06 | FY 07 |
|---|---|---|---|---|---|
| Submissions (PMAs / Panel-track PMA Supplements) |
50 (43/7) |
48 (40/8) |
58 (46/12) |
56 (40/16) |
36 (33/3) |
| FY 2007 Amendments (major deficiency / approvable) |
1 (0/1) |
0 (0/0) |
10 (7/3) |
32 (23/9) |
5 (5/0) |
| Total Amendments (major deficiency / approvable) |
28 (25/3) |
30* (27*/3*) |
55 (44/11) |
36 (27/9) |
5 (5/0) |
* FY 2004 number was updated to reflect a correction to the amendments reported in the FY 2006 MDUFMA Performance Report.
Decisions. Preliminary results for the FY 2006 and FY 2007 cohorts indicate FDA is exceeding the MDUFMA performance goals for making an “FDA decision” (see table below). FDA made decisions on almost three-fourths (40 of 56) of the FY 2006 cohort and one-sixth (6 of 36) of the FY 2007 cohort.
| Goals | Review Within | Fiscal Year |
Cohort Closed |
Number on Time / Actions Completed |
Percent on Time | MDUFMA Performance Goal |
|---|---|---|---|---|---|---|
| Make an "FDA decision" | 320 days | 2003 | N | 45 / 49 | 92% | No Goal |
| 2004 | Y | 41*/ 48 | 85%* | No Goal | ||
| 2005 | N | 48 / 54 | 89% | No Goal | ||
| 2006 | N | 37 / 40 | 93% | 80% | ||
| 2007 | N | 6 / 6 | 100% | 90% | ||
| 180 days | 2007 | N | 6 / 6 | 100% | 50% |
* FY 2004 numbers were updated to reflect corrections to on-time actions reported in the FY 2006 MDUFMA Performance Report.
First Action Letters. With all actions completed for the FY 2005 and FY 2006 cohorts, FDA exceeded the MDUFMA performance goals for issuing first action letters (see table below). Preliminary results indicate that with almost three-fourths (26 of 36) of the FY 2007 cohort completed, FDA is exceeding the MDUFMA performance goals.
| Goals | Review Within | Fiscal Year |
Cohort Closed |
Number on Time / Actions Completed |
Percent on Time | MDUFMA Performance Goal |
|---|---|---|---|---|---|---|
| Issue a "major deficiency" letter as the first action |
150 days | 2003 | Y | 22 / 26 | 85% | No Goal |
| 2004 | Y | 23 / 28 | 82% | No Goal | ||
| 2005 | Y | 34 / 37 | 92% | 75% | ||
| 2006 | Y | 29 / 33 | 88% | 80% | ||
| 2007 | N | 20 / 22 | 91% | 90% | ||
| Issue all other first actions | 180 days | 2003 | Y | 23 / 24 | 96% | No Goal |
| 2004 | Y | 19 / 20 | 95% | No Goal | ||
| 2005 | Y | 19 / 21 | 91% | 75% | ||
| 2006 | Y | 19 / 23 | 83% | 80% | ||
| 2007 | N | 4 / 4 | 100% | 90% |
Second or Later Actions. Preliminary results for the FY 2005 and FY 2006 cohorts indicate the level of FDA performance is below the MDUFMA performance goals for issuing second or later actions (see table below). As of September 30, 2007, FDA had not completed any second or later actions for the FY 2007 cohort.
| Goals | Review Within | Fiscal Year |
Cohort Closed |
Number on Time / Actions Completed |
Percent on Time | MDUFMA Performance Goal |
|---|---|---|---|---|---|---|
| Issue a "major deficiency" letter as the second or later action | 120 days | 2003 | N | 2 / 2 | 100% | No Goal |
| 2004 | N | 4 / 4 | 100% | No Goal | ||
| 2005 | N | 13 / 18 | 72% | 75% | ||
| 2006 | N | 9 / 12 | 75% | 80% | ||
| 2007 | N | 0 / 0 | n/a | 90% |
Amendments to “Major Deficiency” or “Not Approvable” Letters. Preliminary results for the FY 2005 through FY 2007 cohorts indicate FDA is exceeding the MDUFMA performance goal for acting on amendments containing complete responses to “major deficiency” or “not approvable” letters (see table below). FDA reviewed and acted on all amendments received in FY 2007 for the FY 2003 through FY 2007 cohorts.
| Goals | Review Within | Fiscal Year |
Cohort Closed |
Number on Time / Actions Completed |
Percent on Time | MDUFMA Performance Goal |
|---|---|---|---|---|---|---|
| Act on an amendment containing a complete response to a "major deficiency" or "not approvable" letter |
180 days | 2003 | N | 23 / 25 | 92% | No Goal |
| 2004 | N | 21 / 27 | 78% | No Goal | ||
| 2005 | N | 38 / 44 | 86% | 75% | ||
| 2006 | N | 23 / 27 | 85% | 80% | ||
| 2007 | N | 5 / 5 | 100% | 90% |
Amendments to “Approvable” Letters. Preliminary results for the FY 2003 through FY 2006 cohorts indicate FDA is not meeting the MDUFMA performance goals for acting on amendments containing complete responses to “approvable” letters (see table below). However, due to the small number of completed actions on these cohorts, a single additional action will significantly change the on-time performance level. FDA reviewed and acted on all amendments received in FY 2007 for the FY 2003 through FY 2007 cohorts.
| Goals | Review Within | Fiscal Year |
Cohort Closed |
Number on Time / Actions Completed |
Percent on Time | MDUFMA Performance Goal |
|---|---|---|---|---|---|---|
| Act on an amendment containing a complete response to an "approvable" letter |
30 days | 2003 | N | 1 / 3 | 33% | 90% |
| 2004 | N | 0 / 3 | 0% | 90% | ||
| 2005 | N | 8 / 11 | 73% | 90% | ||
| 2006 | N | 5 / 9 | 56% | 90% | ||
| 2007 | N | 0 / 0 | n/a | 90% |
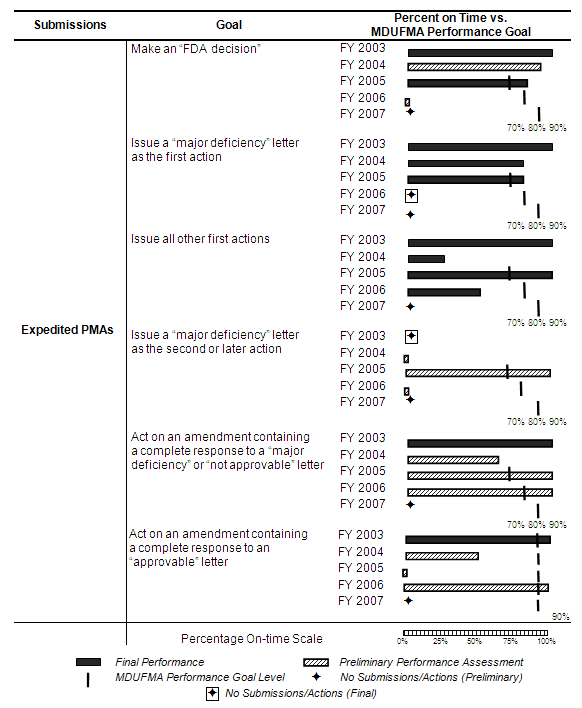
The table below summarizes the annual review time goals for expedited PMAs. In FY 2003, one cycle goal (30 days) became effective. A decision goal and four additional cycle goals became effective in FY 2005 with the performance levels increasing incrementally from 70 percent in FY 2005 to 90 percent in FY 2007.
| Goals | Review Time Goal (Days) | Performance Level | |||||
|---|---|---|---|---|---|---|---|
| FY 03 | FY 04 | FY 05 | FY 06 | FY 07 | |||
| Decision | Make an "FDA decision" | 300 | No Goal | 70% | 80% | 90% | |
| Cycle | Issue a "major deficiency" letter as the first action |
120 | No Goal | 70% | 80% | 90% | |
| Issue all other first actions | 170 | No Goal | 70% | 80% | 90% | ||
| Issue a "major deficiency" letter as the second or later action |
100 | No Goal | 70% | 80% | 90% | ||
| Act on an amendment containing a complete response to a "major deficiency" or "not approvable" letter |
170 | No Goal | 70% | 80% | 90% | ||
| Act on an amendment containing a complete response to an "approvable" letter |
30 | 90% | |||||
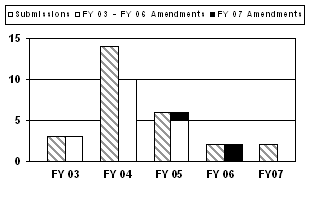
Three out of the last 5 years had three or less expedited PMA submissions, including the last 2 years where two submissions were received in both FY 2006 and FY 2007. Additionally, three amendments were received in FY 2007 with one applying to the FY 2005 cohort and two to the FY 2006 cohort (see corresponding graph and table).
| Expedited PMAs | |||||
|---|---|---|---|---|---|
| Type | FY 03 | FY 04 | FY 05 | FY 06 | FY 07 |
| Submissions | 3 | 14 | 6 | 2 | 2 |
| FY 2007 Amendments (major deficiency / approvable) |
0 (0/0) |
0 (0/0) |
1 (0/1) |
2 (1/1) |
0 (0/0) |
| Total Amendments (major deficiency / approvable) |
3 (2/1) |
10 (8/2) |
6 (5/1) |
2 (1/1) |
0 (0/0) |
Decisions. With all actions completed for the FY 2005 cohort, FDA has exceeded the MDUFMA performance goal for making an “FDA decision” (see table below). FDA made a decision on one of two total submissions for the FY 2006 cohort. With one submission pending, FDA will not meet the FY 2006 MDUFMA performance goal. As of September 30, 2007, FDA had not made decisions on any submissions for the FY 2007 cohort.
| Goals | Review Within | Fiscal Year |
Cohort Closed |
Number on Time / Actions Completed |
Percent on Time | MDUFMA Performance Goal |
|---|---|---|---|---|---|---|
| Make an "FDA decision" | 300 days | 2003 | Y | 3 / 3 | 100% | No Goal |
| 2004 | N | 12 / 13 | 92% | No Goal | ||
| 2005 | Y | 5 / 6 | 83% | 70% | ||
| 2006 | N | 0 / 1 | 0% | 80% | ||
| 2007 | N | 0 / 0 | n/a | 90% |
First Action Letters. All actions were completed for the FY 2005 and FY 2006 cohorts (see table below). FDA exceeded the MDUFMA performance goals for issuing first action letters for the FY 2005 cohorts. FDA did not issue one of two first action letters on time for the FY 2006 cohort, which resulted in FDA not meeting the MDUFMA performance goal. As of September 30, 2007, FDA had not issued any first action letters for the FY 2007 cohort.
| Goals | Review Within | Fiscal Year |
Cohort Closed |
Number on Time / Actions Completed |
Percent on Time | MDUFMA Performance Goal |
|---|---|---|---|---|---|---|
| Issue a "major deficiency" letter as the first action |
120 days | 2003 | Y | 2 / 2 | 100% | No Goal |
| 2004 | Y | 8 / 10 | 80% | No Goal | ||
| 2005 | Y | 4 / 5 | 80% | 70% | ||
| 2006 | Y | 0 / 0 | n/a | 80% | ||
| 2007 | N | 0 / 0 | n/a | 80% | ||
| Issue all other first actions | 170 days | 2003 | Y | 1 / 1 | 100% | No Goal |
| 2004 | Y | 1 / 4 | 25% | No Goal | ||
| 2005 | Y | 1 / 1 | 100% | 70% | ||
| 2006 | Y | 1 / 2 | 50% | 80% | ||
| 2007 | N | 0 / 0 | n/a | 90% |
Second or Later Actions. Preliminary results for the FY 2005 cohort indicate FDA is exceeding the MDUFMA performance goal for issuing second or later actions (see table below). For the FY 2006 cohort, preliminary results indicate that FDA is not meeting the MDUFMA performance goal. However, due to the small number of completed actions on these cohorts, a single additional action will significantly change the on-time performance level. As of September 30, 2007, FDA had not completed any second or later actions for the FY 2007 cohort.
| Goals | Review Within | Fiscal Year |
Cohort Closed |
Number on Time / Actions Completed |
Percent on Time | MDUFMA Performance Goal |
|---|---|---|---|---|---|---|
| Issue a "major deficiency" letter as the second or later action | 100 days | 2003 | Y | 0 / 0 | n/a | No Goal |
| 2004 | N | 0 / 1 | 0% | No Goal | ||
| 2005 | N | 2 / 2 | 100% | 70% | ||
| 2006 | N | 0 / 1 | 0% | 80% | ||
| 2007 | N | 0 / 0 | n/a | 90% |
Amendments to “Major Deficiency” or “Not Approvable” Letters. Preliminary results for the FY 2005 and FY 2006 cohorts indicate FDA is exceeding the MDUFMA performance goals for acting on amendments containing complete responses to “major deficiency” or “not approvable” letters (see table below). However, due to the small number of completed actions on these cohorts, a single additional action will significantly change the on-time performance level. FDA reviewed and acted on all amendments received in FY 2007 for the FY 2003 through FY 2007 cohorts.
| Goals | Review Within | Fiscal Year |
Cohort Closed |
Number on Time / Actions Completed |
Percent on Time | MDUFMA Performance Goal |
|---|---|---|---|---|---|---|
| Act on an amendment containing a complete response to a "major deficiency" or "not approvable" letter |
170 days | 2003 | Y | 2 / 2 | 100% | No Goal |
| 2004 | N | 5 / 8 | 63% | No Goal | ||
| 2005 | N | 5 / 5 | 100% | 70% | ||
| 2006 | N | 1 / 1 | 100% | 80% | ||
| 2007 | N | 0 / 0 | n/a | 90% |
Amendments to “Approvable” Letters. Preliminary results indicate FDA is exceeding the MDUFMA performance goal for the FY 2003 cohort for acting on amendments containing complete responses to “approvable” letters, not meeting the MDUFMA performance goal for the FY 2004 and FY 2005 cohorts, and exceeding the MDUFMA performance goal for the FY 2006 cohort (see table below). However, due to the small number of completed actions on these cohorts, a single additional action will significantly change the on-time performance level. FDA reviewed and acted on all amendments received in FY 2007 for the FY 2003 through FY 2007 cohorts.
| Goals | Review Within | Fiscal Year |
Cohort Closed |
Number on Time / Actions Completed |
Percent on Time | MDUFMA Performance Goal |
|---|---|---|---|---|---|---|
| Act on an amendment containing a complete response to an "approvable" letter |
30 days | 2003 | Y | 1 / 1 | 100% | 90% |
| 2004 | N | 1 / 2 | 50% | 90% | ||
| 2005 | N | 0 / 1 | 0% | 90% | ||
| 2006 | N | 1 / 1 | 100% | 90% | ||
| 2007 | N | 0 / 0 | n/a | 90% |
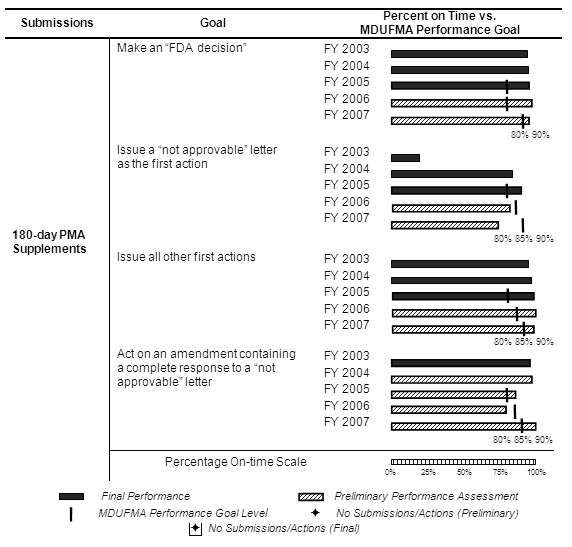
The table below summarizes the annual review time goals for 180-day PMA supplements. The decision goal and three cycle goals became effective in FY 2005 with the performance levels increasing from 80 percent in FY 2005 to 90 percent in FY 2007.
| Goals | Review Time Goal (Days) |
Performance Level | |||||
|---|---|---|---|---|---|---|---|
| FY 03 | FY 04 | FY 05 | FY 06 | FY 07 | |||
| Decision | Make an "FDA decision" | 180 | No Goal | 80% | 80% | 90% | |
| Cycle | Issue a "not approvable" letter as the first action |
120 | No Goal | 80% | 80% | 90% | |
| Issue all other first actions | 180 | No Goal | 80% | 80% | 90% | ||
| Act on an amendment containing a complete response to a "not approvable" letter |
160 | No Goal | 80% | 80% | 90% | ||
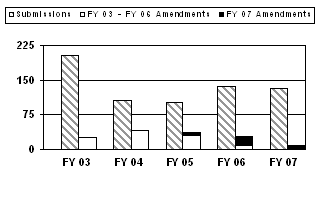
More 180-day PMA supplements were received during FY 2006 and FY 2007 than during the previous 2 years. However, FY 2003 had the highest number of supplements received over the 5-year period. Additionally, 35 amendments were received in FY 2007 with most (21) applying to the FY 2006 cohort (see corresponding graph and table).
180-day PMA Supplements
| Type | FY 03 | FY 04 | FY 05 | FY 06 | FY 07 |
|---|---|---|---|---|---|
| Submissions | 204 | 106 | 101 | 136 | 132 |
| FY 2007 Amendments | 0 | 0 | 6 | 21 | 8 |
| Total Amendments | 25 | 42 | 36 | 29 | 8 |
Decisions. With all actions completed for the FY 2005 cohort, FDA exceeded the MDUFMA performance goal for making an “FDA decision” (see table below). FDA made decisions on all but two (134 of 136) of the FY 2006 cohort and, with only two submissions pending, FDA is assured of exceeding the MDUFMA performance goal. With almost two-thirds (80 of 132) of the FY 2007 cohort completed, preliminary results indicate that FDA is exceeding the MDUFMA performance goal.
| Goals | Review Within | Fiscal Year |
Cohort Closed |
Number on Time / Actions Completed16 |
Percent on Time | MDUFMA Performance Goal |
|---|---|---|---|---|---|---|
| Make an "FDA decision" | 180 days | 2003 | Y | 192 / 204 | 94% | No Goal |
| 2004 | Y | 101* / 106 | 95%* | No Goal | ||
| 2005 | Y | 96 / 101 | 95% | 80% | ||
| 2006 | N | 130 / 134 | 97% | 80% | ||
| 2007 | N | 76 / 80 | 95% | 90% |
* FY 2004 numbers were updated to reflect corrections to on-time actions reported in the FY 2006 MDUFMA Performance Report.
First Action Letters. With all actions completed for the FY 2005 cohort, FDA exceeded the MDUFMA performance goals for issuing first action letters (see table below). With one submission pending (135 of 136) for the FY 2006 cohort, FDA will not meet the MDUFMA performance goal for “not approvable” letters but will exceed the FY 2006 MDUFMA performance goal for issuing all other first actions. Preliminary results for the FY 2007 cohort indicate that, with almost two-thirds (82 of 132) of the cohort completed, FDA is not meeting the MDUFMA performance goal for “not approvable” letters while exceeding the MDUFMA performance goal for issuing all other first actions.
| Goals | Review Within | Fiscal Year |
Cohort Closed |
Number on Time / Actions Completed |
Percent on Time | MDUFMA Performance Goal |
|---|---|---|---|---|---|---|
| Issue a "not approvable" letter as the first action |
120 days | 2003 | Y | 6 / 32 | 19% | No Goal |
| 2004 | Y | 36 / 43 | 84% | No Goal | ||
| 2005 | Y | 36 / 40 | 90% | 80% | ||
| 2006 | N | 33 / 41 | 81% | 85% | ||
| 2007 | N | 17 / 23 | 74% | 90% | ||
| Issue all other first actions | 180 days | 2003 | Y | 164 / 172 | 95% | No Goal |
| 2004 | Y | 61 / 63 | 97% | No Goal | ||
| 2005 | Y | 60 / 61 | 98% | 80% | ||
| 2006 | N | 93 / 94 | 99% | 85% | ||
| 2007 | N | 58 / 59 | 98% | 90% |
Amendments to “Not Approvable” Letters. Preliminary results indicate that FDA is exceeding the MDUFMA performance goal for the FY 2005 cohort for acting on amendments containing complete responses to “not approvable” letters, not meeting the MDUFMA performance goal for the FY 2006 cohort, and exceeding the MDUFMA performance goal for the FY 2007 cohort (see table below). FDA reviewed and acted on all amendments received in FY 2007 for the FY 2003 through FY 2007 cohorts.
| Goals | Review Within | Fiscal Year |
Cohort Closed |
Number on Time / Actions Completed |
Percent on Time | MDUFMA Performance Goal |
|---|---|---|---|---|---|---|
| Act on an amendment containing a complete response to a "not approvable" letter |
160 days | 2003 | Y | 24 / 25 | 96% | No Goal |
| 2004 | N | 41 / 42 | 98% | No Goal | ||
| 2005 | N | 31 / 36 | 86% | 80% | ||
| 2006 | N | 23 / 29 | 79% | 85% | ||
| 2007 | N | 8 / 8 | 100% | 90% |
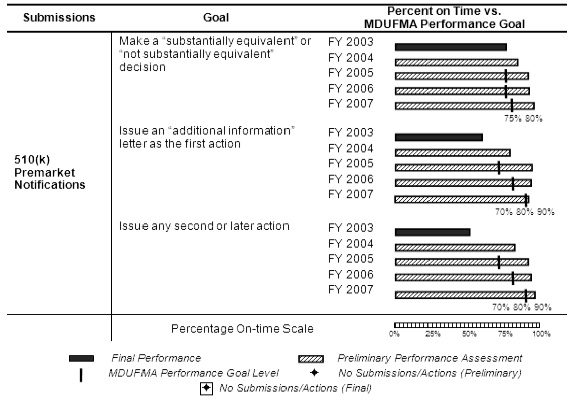
The table below summarizes the annual review time goals for 510(k) premarket notifications. The decision goal and two cycle goals became effective in FY 2005. The performance level for the decision goal remained constant at 75 percent for FY 2005 and FY 2006 and increased to 80 percent in FY 2007. The performance levels for the two cycle goals increased incrementally from 70 percent in FY 2005 to 90 percent in FY 2007.
| Goals | Review Time Goal (Days) |
Performance Level | |||||
|---|---|---|---|---|---|---|---|
| FY 03 | FY 04 | FY 05 | FY 06 | FY 07 | |||
| Decision | Make a "substantially equivalent" or "not substantially equivalent" decision | 90 | No Goal | 75% | 80% | ||
| Cycle | Issue an "additional information" letter as the first action |
75 | No Goal | 70% | 80% | 90% | |
| Issue any second or later action |
60 | No Goal | 70% | 80% | 90% | ||
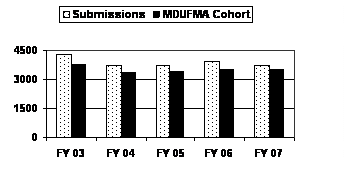
The number of 510(k) submissions received in FY 2007 returned to the FY 2004 and FY 2005 levels after increasing in FY 2006. The MDUFMA cohort portion of 510(k) submissions fluctuated over the same period and reached a 5-year high of 95 percent (3,531 of 3,713) of total submissions in FY 2007 (see corresponding graph and table).5
510(k) Premarket Notifications
| Type | FY 03 | FY 04 | FY 05 | FY 06 | FY 07 |
|---|---|---|---|---|---|
| Submissions | 4,290 | 3,710 | 3,713 | 3,913 | 3,713 |
| MDUFMA Cohort | 3,795 | 3,382 | 3,405 | 3,530 | 3,531 |
Decisions. FDA made decisions on almost all (3,401 of 3,405) of the FY 2005 cohort and almost all (3,466 of 3,530) of the FY 2006 cohort (see table below). With submissions still pending, FDA will exceed the FY 2005 and FY 2006 MDUFMA performance goals. Preliminary results indicate that with almost two-thirds (2,206 of 3,531) of the FY 2007 cohort completed, FDA is exceeding the MDUFMA performance goal.
| Goals | Review Within | Fiscal Year |
Cohort Closed |
Number on Time / Actions Completed |
Percent on Time | MDUFMA Performance Goal |
|---|---|---|---|---|---|---|
| Make a "substantially equivalent" or "not substantially equivalent" decision | 90 days | 2003 | Y | 2,887 / 3,795 | 76% | No Goal |
| 2004 | N | 2,835 / 3,381 | 84% | No Goal | ||
| 2005 | N | 3,100 / 3,401 | 91% | 75% | ||
| 2006 | N | 3,191 / 3,466 | 92% | 75% | ||
| 2007 | N | 2,085 / 2,206 | 95% | 80% |
First Action Letters. Preliminary results for the FY 2005 through FY 2007 cohorts indicate FDA is exceeding the MDUFMA performance goals for issuing first action letters (see table below). FDA issued first action letters for over half (1,847 of 3,405) of the FY 2005 cohort, almost two-thirds (2,174 of 3,530) of the FY 2006 cohort, and over half (1,925 of 3,531) of the FY 2007 cohort.
| Goals | Review Within | Fiscal Year |
Cohort Closed |
Number on Time / Actions Completed |
Percent on Time | MDUFMA Performance Goal |
|---|---|---|---|---|---|---|
| Issue an "additional information" letter as the first action |
75 days | 2003 | Y | 1,011 / 1,726 | 59% | No Goal |
| 2004 | N | 1,271 / 1,618 | 79% | No Goal | ||
| 2005 | N | 1,732 / 1,847 | 94% | 70% | ||
| 2006 | N | 2,017/ 2,174 | 93% | 80% | ||
| 2007 | N | 1,774 / 1,925 | 92% | 90% |
Second or Later Actions. Preliminary results for the FY 2005 through FY 2007 cohorts indicate FDA is exceeding the MDUFMA performance goals for issuing second or later actions (see table below).
| Goals | Review Within | Fiscal Year |
Cohort Closed |
Number on Time / Actions Completed |
Percent on Time | MDUFMA Performance Goal |
|---|---|---|---|---|---|---|
| Issue any second or later action | 60 days | 2003 | Y | 311 / 611 | 51% | No Goal |
| 2004 | N | 480 / 587 | 82% | No Goal | ||
| 2005 | N | 615 / 672 | 92% | 70% | ||
| 2006 | N | 796 / 861 | 93% | 80% | ||
| 2007 | N | 508 / 528 | 96% | 90% |
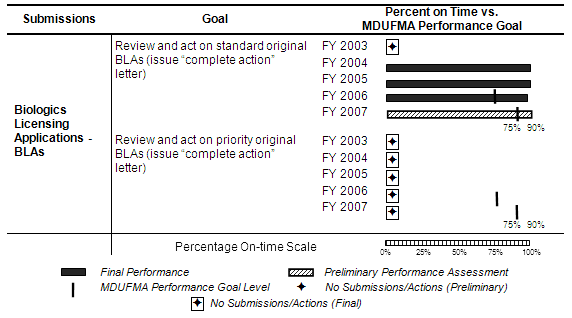
The table below summarizes review time goals for BLAs for FY 2006 MDUFMA performance levels for standard and priority original BLA submissions. Performance levels increased from 75 percent in FY 2006 to 90 percent in FY 2007.
| Goals | Review Time Goal (Months) |
Performance Level | |||||
|---|---|---|---|---|---|---|---|
| FY 03 | FY 04 | FY 05 | FY 06 | FY 07 | |||
| Review and act on standard original BLAs (issue "complete action" letter) | 10 | No Goals | 75% | 90% | |||
| Review and act on priority original BLAs (issue "complete action" letter) | 6 | No Goals | 75% | 90% | |||
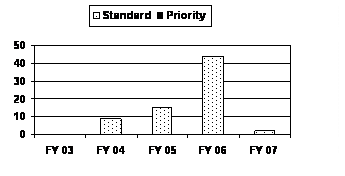
The number of standard BLAs submitted increased from FY 2003 to FY 2006 with the highest increase occurring in FY 2006. Following this increase, the standard BLAs submitted in FY 2007 decreased to a 4-year low. No priority BLAs were received from FY 2003 through FY 2007 (see corresponding graph and table).
Original BLAs
| Type | FY 03 | FY 04 | FY 05 | FY 06 | FY 07 |
|---|---|---|---|---|---|
| Standard | 0 | 9 | 15 | 44 | 2 |
| Priority | 0 | 0 | 0 | 0 | 0 |
| MDUFMA Total | 0 | 9 | 15 | 44 | 2 |
Complete Action Letters. With all standard BLA submissions reviewed and acted on for the FY 2006 cohort, FDA exceeded the MDUFMA performance goal (see table below). Preliminary results indicate that with only one standard BLA submission reviewed and acted on for the FY 2007 cohort, FDA is exceeding the MDUFMA performance goal. However, action on the one submission pending for the FY 2007 cohort could change the on-time performance level. With no submissions received for priority BLAs for FY 2006 and FY 2007, on-time performance is not applicable.
| Goals | Review Within | Fiscal Year |
Cohort Closed |
Number on Time / Actions Completed |
Percent on Time | MDUFMA Performance Goal |
|---|---|---|---|---|---|---|
| Review and act on standard original BLAs (issue "complete action" letter) |
10 months | 2003 | Y | 0 / 0 | n/a | No Goal |
| 2004 | Y | 9 / 9 | 100% | No Goal | ||
| 2005 | Y | 15 / 15 | 100% | No Goal | ||
| 2006 | Y | 43 / 44 | 98% | 75% | ||
| 2007 | N | 1 / 1 | 100% | 90% | ||
| Review and act on priority original BLAs (issue "complete action" letter) |
6 months | 2003 | Y | 0 / 0 | n/a | No Goal |
| 2004 | Y | 0 / 0 | n/a | No Goal | ||
| 2005 | Y | 0 / 0 | n/a | No Goal | ||
| 2006 | Y | 0 / 0 | n/a | 75% | ||
| 2007 | Y | 0 / 0 | n/a | 90% |
The table below summarizes annual review time goals for BLA Supplements for FY 2006 MDUFMA performance levels for standard and priority BLA efficacy supplements and BLA manufacturing supplements. Performance levels increased from 75 percent in FY 2006 to 90 percent in FY 2007.
| Goals | Review Time Goal (Months) |
Performance Level | ||||
|---|---|---|---|---|---|---|
| FY 03 | FY 04 | FY 05 | FY 06 | FY 07 | ||
| Review and act on standard BLA efficacy supplements (issue "complete action" letter) | 10 | No Goals | 75% | 90% | ||
| Review and act on priority BLA efficacy supplements (issue "complete action" letter) | 6 | No Goals | 75% | 90% | ||
| Review and act on BLA manufacturing supplements that require prior approval (issue "complete action" letter) | 4 | No Goals | 75% | 90% | ||
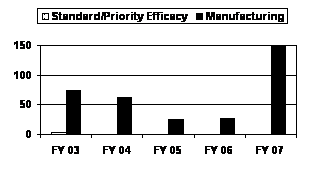
The number of BLA manufacturing supplements increased almost sixfold in FY 2007 from FY 2005 and FY 2006 levels, reaching a 5-year high. No standard or priority BLA efficacy supplements were submitted in the past 4 years (see corresponding graph and table).
BLA Supplements
| Type | FY 03 | FY 04 | FY 05 | FY 06 | FY 07 |
|---|---|---|---|---|---|
| Standard Efficacy | 3 | 0 | 0 | 0 | 0 |
| Priority Efficacy | 0 | 0 | 0 | 0 | 0 |
| Manufacturing | 73 | 62 | 25 | 26 | 148 |
| MDUFMA Total | 76 | 62 | 25 | 26 | 148 |
Complete Action Letters. With all BLA manufacturing supplements for the FY 2006 cohort reviewed and acted on, FDA exceeded the MDUFMA performance goal (see table below). FDA reviewed and acted on almost all (134 of 148) BLA manufacturing supplements for the FY 2007 cohort and with submissions pending, FDA is assured of exceeding the MDUFMA performance goal. With no submissions received for BLA standard and priority efficacy supplements for FY 2006 and FY 2007, on-time performance is not applicable.
| Goals | Review Within | Fiscal Year |
Cohort Closed |
Number on Time / Actions Completed |
Percent on Time | MDUFMA Performance Goal |
|---|---|---|---|---|---|---|
| Review and act on standard BLA efficacy supplements (issue "complete action" letter) | 10 months | 2003 | Y | 3 / 3 | 100% | No Goal |
| 2004 | Y | 0 / 0 | n/a | No Goal | ||
| 2005 | Y | 0 / 0 | n/a | No Goal | ||
| 2006 | Y | 0 / 0 | n/a | 75% | ||
| 2007 | Y | 0 / 0 | n/a | 90% | ||
| Review and act on priority BLA efficacy supplements (issue "complete action" letter) |
6 months | 2003 | Y | 0 / 0 | n/a | No Goal |
| 2004 | Y | 0 / 0 | n/a | No Goal | ||
| 2005 | Y | 0 / 0 | n/a | No Goal | ||
| 2006 | Y | 0 / 0 | n/a | 75% | ||
| 2007 | Y | 0 / 0 | n/a | 90% | ||
| Review and act on BLA manufacturing supplements that require prior approval (issue "complete action" letter) |
4 months | 2003 | Y | 72 / 73 | 99% | No Goal |
| 2004 | Y | 62 / 62 | 100% | No Goal | ||
| 2005 | Y | 24 / 25 | 96% | No Goal | ||
| 2006 | Y | 26 / 26 | 100% | 75% | ||
| 2007 | N | 134 / 134 | 100% | 90% |
The table below summarizes the annual review time goals for resubmitted original BLAs and BLA efficacy supplements for the “Class 1” and “Class 2” resubmissions. Performance levels increased incrementally from 75 percent in FY 2005 to 90 percent in FY 2007.
| Goals | Review Time Goal (Months) |
Performance Level | ||||
|---|---|---|---|---|---|---|
| FY 03 | FY 04 | FY 05 | FY 06 | FY 07 | ||
| Review and act on "Class 1" original BLA and BLA efficacy supplement resubmissions | 2 | No Goals | 75% | 80% | 90% | |
| Review and act on "Class 2" original BLA and BLA efficacy supplement resubmissions | 6 | No Goals | 75% | 80% | 90% | |
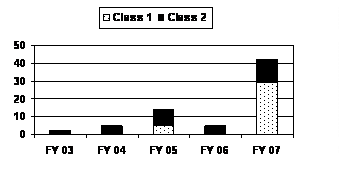
The number of resubmitted BLAs and BLA efficacy supplements increased over eightfold in FY 2007, reaching a 5-year high for both “Class 1” and “Class 2” resubmissions. FDA received no “Class 1” resubmissions and five or less “Class 2” resubmissions in three of the last 5 years (see corresponding graph and table).
Resubmitted BLAs and BLA Efficacy Supplements
| Type | FY 03 | FY 04 | FY 05 | FY 06 | FY 07 |
|---|---|---|---|---|---|
| “Class 1” | 0 | 0 | 5 | 0 | 29 |
| “Class 2” | 2 | 5 | 9 | 5 | 13 |
| MDUFMA Total | 2 | 5 | 14 | 5 | 42 |
Resubmissions. With all “Class 1” and “Class 2” BLA and BLA efficacy supplement resubmissions reviewed and acted on for FY 2005 through FY 2007 cohorts, FDA exceeded the MDUFMA performance goals for all three fiscal year cohorts (see table below). With no “Class 1” BLA and BLA efficacy supplement resubmissions received for FY 2006, on-time performance is not applicable.
| Goals | Review Within | Fiscal Year |
Cohort Closed |
Number on Time / Actions Completed |
Percent on Time | MDUFMA Performance Goal |
|---|---|---|---|---|---|---|
| Review and act on "Class 1" original BLA and BLA efficacy supplement resubmissions | 10 months | 2003 | Y | 0 / 0 | n/a | No Goal |
| 2004 | Y | 0 / 0 | n/a | No Goal | ||
| 2005 | Y | 5 / 5 | 100% | 75% | ||
| 2006 | Y | 0 / 0 | n/a | 80% | ||
| 2007 | Y | 29 / 29 | 100% | 90% | ||
| Review and act on "Class 2" original BLA and BLA efficacy supplement resubmissions | 6 months | 2003 | Y | 2 / 2 | 100% | No Goal |
| 2004 | Y | 4 / 5 | 80% | No Goal | ||
| 2005 | Y | 9 / 9 | 100% | 75% | ||
| 2006 | Y | 5 / 5 | 100% | 80% | ||
| 2007 | Y | 13 / 13 | 100% | 90% |
This section reports on the additional commitments outlined in FDA’s Commitment Letter. A detailed description of the commitments, performance targets, and definitions of terms can be found in Appendix A (section I, paragraphs I - P).
FDA’s FY 2007 review performance for submissions that do not have specific MDUFMA performance goals continued to be comparable to FY 2002 performance (prior to enactment of MDUFMA).
| CDRH Performance Indicators | FY 02 | FY 03 | FY 04 | FY 05 | FY 06 | FY 06 |
|---|---|---|---|---|---|---|
| HDEs - Filing to first action (average FDA days) | 53 | 48 | 52 | 63 | 67 | 79 |
| HDEs - Elapsed time to approval (average FDA days) | 175 | 152 | 182 | 223 | 297 | 230 |
| IDEs - FDA review time (average FDA days) | 28 | 27 | 28 | 28 | 28 | 27 |
| IDEs - Percent of decisions made within 30 days | 99% | 100% | 100% | 96% | 99% | 99% |
| IDE Amendments - FDA review time (average FDA days) | 18 | 19 | 18 | 20 | 19 | 20 |
| IDE Amendments - Percent of decisions made within 30 days | 100% | 100% | 100% | 98% | 100% | 98% |
| IDE Supplements - FDA review time (average FDA days) | 20 | 19 | 19 | 19 | 20 | 21 |
| IDE Supplements - Percent of decisions made within 30 days | 100% | 100% | 100% | 100% | 100% | 99% |
| CBER Performance Indicators | FY 02 | FY 03 | FY 04 | FY 05 | FY 06 | FY 06 |
| BLA Supplements (CBE/CBE-30) - Percent reviewed and acted on within 6 months | 99% | 97% | 100% | 100% | 100% | 100% |
| PMA Supplements (CBE) - Percent of decisions made within 180 days | 100% | 100% | 100% | 100% | 100% | 100% |
| PMA Supplements (135-day) - Percent of decisions made within 135 days | NR | 100% | 100% | 100% | 100% | 100% |
| PMA Supplements (CBE-30) - Percent of decisions made within 30 days | 67% | 100% | 100% | 100% | 100% | 100% |
| KEY: HDEs-Humanitarian Device Exemptions; IDEs-Investigational Device Exemptions; BLA-Biologics Licensing Application; PMA-Premarket Application; CBE-Changes Being Effected; NR-None Received |
||||||
NOTE: Some reported measures may change over time, as additional actions are taken on open applications.
FDA continues to encourage meetings with regulated industry as an effective way to ensure that both FDA and applicants understand the clinical, scientific, and regulatory issues associated with new technologies. Pre-IDE and pre-PMA meetings have shown to be beneficial and are used routinely by industry. During FY 2007, FDA participated in more than 1,500 premarket meetings with industry. No pre-PMA meeting requests were received in FY 2007 for CBER. The more formal types of meetings (agreement, determination, and 100-day meetings) are not used as frequently by premarket applicants.
FDA’s annual financial Reports to Congress provide information on FDA’s use of resources for the MDUFMA program and are available at: http://www.fda.gov/oc/mdufma/.
Modular PMA Review Program
FDA issued initial guidance on modular PMA reviews in its guidance document, Assessing User Fees: PMA Supplement Definitions, Modular PMA Fees, BLA and Efficacy Supplement Definitions, Bundling Multiple Devices in a Single Application, and Fees for Combination Products, on February 25, 2003, available at: http://www.fda.gov/cdrh/mdufma/guidance/1201.html. This guidance explains that the fee for a modular PMA submission is due upon submission of the first module (not just the “shell” that describes the overall plan for the modular submission).
On November 23, 2003, FDA provided a more comprehensive guidance document, Premarket Approval Application Modular Review, available at: http://www.fda.gov/cdrh/mdufma/guidance/835.html. This guidance provided industry and FDA staff with information regarding the modular review program and outlined the procedures for submitting and reviewing a modular PMA. As FDA gains more experience with the modular PMA process, it will consult with stakeholders to develop performance goals for this program.
Although FDA extended the modular review program to panel-track PMA supplements, as of the close of FY 2007, FDA had not received a modular panel-track PMA supplement.
After consulting with stakeholders, FDA determined that bundling is appropriate under certain circumstances.6 On February 25, 2003, FDA issued an initial guidance document, Assessing User Fees: PMA Supplement Definitions, Modular PMA Fees, BLA and Efficacy Supplement Definitions, Bundling Multiple Devices in a Single Application, and Fees for Combination Products, describing general bundling principles. This guidance document is available at: http://www.fda.gov/cdrh/mdufma/guidance/1201.html. This guidance explained that bundling may involve multiple devices or multiple indications for use in a single submission. On November 26, 2003, FDA issued a more comprehensive guidance document, Bundling Multiple Devices or Multiple Indications in a Single Submission. FDA published an updated edition of this guidance on June 22, 2007, and it is available at: http://www.fda.gov/cdrh/mdufma/guidance/1215.pdf. This guidance is intended to help industry and FDA staff understand when bundling may be appropriate and when separate submissions should be considered. It also provides numerous examples illustrating these bundling principles for both 510(k) and PMA applications. Interest in bundling has increased since MDUFMA was enacted, and FDA is now receiving more bundled submissions.
FDA published Guidance for Industry, Providing Regulatory Submissions to CBER in Electronic Format - Investigational New Drug Applications (INDs) (March 26, 2002), which applies to investigational studies of devices, such as blood screening test kits, leading to a BLA, available at: http://www.fda.gov/cber/gdlns/eind.htm. CBER contributed to guidance documents on electronic submissions in general, and received a number of electronic submissions for biologic (non-device) reviews. To date, CBER has not received electronic submissions of any medical device applications.
CBER continues to make a significant outreach effort to inform regulated industry of the process for electronic submissions. In particular, during all sponsor meetings, CBER informs applicants and potential applicants of the ability to submit electronic documents. In addition, CBER is making provisions for secure e-mail when not associated with an original electronic application.
CDRH is working with applicants to expand the use of electronic submissions, focusing first on increasing the use of electronic copies of applications. CDRH has initiated a “Turbo 510(k)” pilot, providing an electronic template for submission and review of
in vitro diagnostic device 510(k)s, and will use the experience gained and lessons learned from this pilot as it moves forward with additional electronic initiatives. In FY 2007, industry submitted 49 Turbo 510(k) electronic submissions (compared to 85 in FY 2006). No new electronic templates for radiation safety reports required of electronic product manufacturers were developed in FY 2007 (compared to 24 in FY 2006). In FY 2007, 473 radiological health stakeholders downloaded the applicable software (compared to 448 in FY 2006) and 334 electronic radiation safety reports were submitted by the electronic product industry (compared to 123 in FY 2006).7
During FY 2003, FDA began a comprehensive examination of factors affecting the timeliness and efficiency of the preapproval inspection process to determine how the process can be improved and what resources would be required to make those improvements. In FY 2006, FDA issued guidance that: 1) helps industry better understand the preapproval inspection process, so they will be better prepared for their inspections; and 2) explains how the Centers will work with applicants, the Office of Regulatory Affairs, and with its field inspectors to improve the timeliness of preapproval inspections.
THE SECRETARY OF HEALTH AND HUMAN SERVICES
Washington, DC, November 14, 2002
The Honorable Edward Kennedy
United States Senate
Washington, DC
Dear Mr. Chairman:
As you are aware, the Medical Device User Fee and Modernization Act of 2002 was signed by the President on October 26, 2002. Under Title I, the additional revenues generated from fees paid by the medical device industry will be used to expedite the medical device review process, in accordance with performance goals that were developed by the Food and Drug Administration (FDA) in consultation with the industry.
FDA has worked with various stakeholders, including representatives from consumer, patient, and health provider groups, and the medical device industry to develop legislation and goals that would enhance the success of the device review program. Title I of the Medical Device User Fee and Modernization Act of 2002 reflects the fee mechanisms and other improvements developed in these discussions. The performance goals referenced in Section 101 are specified in the enclosure to this letter, entitled “Performance Goals and Procedures.” I believe they represent a realistic projection of what FDA can accomplish with industry cooperation and the additional resources identified in the bill.
This letter and the enclosed goals document pertain only to title I (Fees Related to Medical Devices) of Public Law 107-250, Medical Device User Fee and Modernization Act of 2002. OMB has advised that there is no objection to the presentation of these views from the standpoint of the Administration’s program. We appreciate the support of you and your staffs, the assistance of other Members of the Committee, and that of the Appropriations Committees, in the authorization of this vital program.
Sincerely,
Tommy G. Thompson
Enclosure
The performance goals and procedures of the FDA Center for Devices and Radiological Health (CDRH) and the Center for Biologics Evaluation and Research (CBER), as agreed to under the medical device user fee program in the Medical Device User Fee and Modernization Act of 2002, are summarized as follows:
I. REVIEW PERFORMANCE GOALS - FISCAL YEAR 2003 THROUGH 2007
All references to "days" mean "FDA days."
A. ORIGINAL PREMARKET APPROVAL (PMA), PANEL-TRACK PMA SUPPLEMENT, AND PREMARKET REPORT SUBMISSIONS
1. The following cycle goals apply to: 75 percent of submission received in fiscal year 2005; 80 percent of submissions received in fiscal year 2006; 90 percent of submissions received in fiscal year 2007.
(a) First action major deficiency letters will issue within 150 days.
(b) All other first action letters (approval, approvable, approvable pending good manufacturing practices (GMP) inspection, not approvable, or denial) will issue within 180 days.
(c) Second or later action major deficiency letters will issue within 120 days.
(d) Amendments containing a complete response to major deficiency or not approvable letters will be acted on within 180 days.
2. Decision Goals:
(a) 80 percent of submissions received in fiscal year 2006 will have an FDA decision in 320 days.
(b) 90 percent of submissions received in fiscal year 2007 will have an FDA decision in 320 days.
3. Subject to the following paragraph, 50 percent of submissions received in fiscal year 2007 will have an FDA decision in 180 days.
This goal will be re-evaluated following the end of fiscal year 2005. FDA will hold a public meeting to consult with its stakeholders and to determine whether this goal is appropriate for implementation in fiscal year 2007. If FDA determines that the goal is not appropriate, prior to August 1, 2006, the Secretary will send a letter to the Committee on Health, Education, Labor and pensions of the Senate and to the Energy and Commerce Committee, Subcommittee on Health of the House of Representatives stating that the goal will not be implemented and the rationale for its removal.
4. 90 percent of amendments containing a complete response to an approvable letter received in fiscal years 2003 through 2007 will be acted on within 30 days.
B. EXPEDITED ORIGINAL PMA SUBMISSIONS
1. The following goals apply to PMA submissions where:
(a) FDA has granted the application expedited status;
(b) The applicant has requested and attended a pre-filing review meeting with FDA;
(c) The applicant’s manufacturing facilities are prepared for inspection upon submission of the application; and
(d) The application is substantively complete, as defined at the pre-filing review meeting.
2. The following cycle goals apply to: 70 percent of submissions received in fiscal year 2005; 80 percent of submissions received in fiscal year 2006; 90 percent of submissions received in fiscal year 2007.
(a) First action major deficiency letters will issue within 120 days.
(b) All other first action letters (approval, approvable, approvable pending GMP inspection, not approvable, or denial) will issue within 170 days.
(c) Second or later action major deficiency letters will issue within 100 days.
(d) Amendments containing a complete response to major deficiency or not approvable letters will be acted on within 170 days.
3. Decision Goals:
(a) 70 percent of submissions received in fiscal year 2005 will have an FDA decision in 300 days.
(b) 80 percent of submissions received in fiscal year 2006 will have an FDA decision in 300 days.
(c) 90 percent of submissions received in fiscal year 2007 will have an FDA decision in 300 days.
4. 90 percent of amendments containing a complete response to an approvable letter received in fiscal years 2003 through 2007 will be acted on within 30 days.
C. 180-DAY PMA SUPPLEMENT SUBMISSIONS
1. The following goals apply to: 80 percent of submissions in fiscal year 2005; 85 percent of submissions in fiscal year 2006; 90 percent of submissions in fiscal year 2007.
(a) First action not approvable letters will issue within 120 days.
(b) All other first action letters (approval, approvable, approvable pending GMP inspection, or denial) will issue within 180 days. [8]
(c) Amendments containing a complete response to a not approvable letter will be acted on within 160 days.
2. Decision Goals:
(a) 80 percent of submissions received in fiscal year 2005 will have an FDA decision in 180 days.
(b) 80 percent of submissions received in fiscal year 2006 will have an FDA decision in 180 days.
(c) 90 percent of submissions received in fiscal year 2007 will have an FDA decision in 180 days.
3. Current performance for real-time review PMA supplement submissions will be maintained.
D. 510(k) SUBMISSIONS
1. The following goals apply to: 70 percent of submissions received in fiscal year 2005; 80 percent of submissions received in fiscal year 2006; 90 percent of submissions received in fiscal year 2007.
(a) First action additional information letters will issue within 75 days.
(b) Subsequent action letters will issue within 60 days.
2. Decision Goals:
(a) 75 percent of submissions received in fiscal years 2005 and 2006 will have an FDA decision in 90 days.
3. Subject to the following paragraph, 80 percent of submissions received in fiscal year 2007 will have an FDA decision in 90 days.
This goal will be re-evaluated following the end of fiscal year 2005. FDA will hold a public meeting to consult with its stakeholders and to determine whether this goal is appropriate for implementation in fiscal year 2007. If FDA determines that the goal is not appropriate, prior to August 1, 2006, the Secretary will send a letter to the Committee on Health, Education, Labor and Pensions of the Senate and to the Energy and Commerce Committee, Subcommittee on Health of the House of Representatives stating that the goal will not be implemented and the rationale for its removal, and that the goal for fiscal year 2006 will be implemented for fiscal year 2007.
E. ORIGINAL BIOLOGICS LICENSING APPLICATIONS (BLAs)
The following goals apply to: 75 percent of submissions received in fiscal year 2006; 90 percent of submissions received in fiscal year 2007.
1. Review and act on standard original BLA submissions within 10 months of receipt.
2. Review and act on priority original BLA submissions within 6 months of receipt.
F. BLA EFFICACY SUPPLEMENTS
The following goals apply to: 75 percent of submissions received in fiscal year 2006; 90 percent of submissions received in fiscal year 2007.
1. Review and act on standard BLA efficacy supplement submissions within 10 months of receipt.
2. Review and act on priority BLA efficacy supplement submissions within 6 months of receipt.
G. ORIGINAL BLA AND BLA EFFICACY SUPPLEMENT RESUBMISSIONS
The following goals apply to: 75 percent of submissions received in fiscal year 2005; 80 percent of submissions received in fiscal year 2006; 90 percent of submissions received in fiscal year 2007.
1. Review and act on "Class 1" original BLA and BLA efficacy supplement resubmissions within 2 months of receipt.
2. Review and act on "Class 2" original BLA and BLA efficacy supplement resubmissions within 6 months of receipt.
H. BLA MANUFACTURING SUPPLEMENTS REQUIRING PRIOR APPROVAL
The following goal applies to: 75 percent of submissions received in fiscal year 2006; 90 percent of submissions received in fiscal year 2007.
Review and act on BLA manufacturing supplements requiring prior approval within 4 months of receipt.
I. ADDITIONAL EFFORTS RELATED TO PERFORMANCE GOALS
The Agency and the regulated industry agree that the use of both informal and formal meetings (e.g., determination and agreement meetings, informal pre-investigational device exemption (IDE) meetings, pre-PMA meetings, pre-PMA filing meetings) by both parties is critical to ensure high application quality such that the above performance goals can be achieved.
J. MAINTENANCE OF CURRENT PERFORMANCE
It is the intent of the Agency that in review areas where specific performance goals have not been identified, current performance will be maintained.
K. APPLICATION OF USER FEE REVENUES
The Agency intends to apply significant user fee revenues to support reviewer training and hiring and/or outside contracting to achieve the identified performance goals in a responsible and efficient manner.
L. MODULAR PMA REVIEW PROGRAM
The Agency intends to issue guidance regarding the implementation of new section 515(c)(3) of the Federal Food, Drug, and Cosmetic Act. It is the intent of the Agency that once this program is implemented, the Agency will work with its stakeholders to develop appropriate performance goals for this program. Until such time, the Agency intends to review and close complete modules that are submitted well in advance of the PMA submission as expeditiously as possible.
M. "FOLLOW-ON" LICENSED DEVICES
The Center for Biologics Evaluation and Research will, if feasible, identify a category of "follow-on" licensed devices and collect information to determine whether alternative performance goals for such a category are appropriate.
N. BUNDLING POLICY
The Agency will, in consultation with its stakeholders, consider the issue of bundling for products with multiple related submissions. After such consultation, the Agency will either issue guidance on bundling or publish a notice explaining why it has determined that bundling is inappropriate.
O. ELECTRONIC REVIEW OF APPLICATIONS
The Agency will continue its efforts toward development of electronic receipt and review of applications, as expeditiously as possible, acknowledging that insufficient funding is included in the user fee program for this effort.
P. PREAPPROVAL INSPECTIONS
The Agency will plan to improve the scheduling and timeliness of preapproval inspections. The Agency will monitor the progress of these efforts and provide such information in the annual performance report.
II. ANNUAL STAKEHOLDER MEETING
Beginning in fiscal year 2004, FDA will hold annual public meetings to review and evaluate the implementation of this program in consultation with its stakeholders.
III. DEFINITIONS AND EXPLANATION OF TERMS
A. For original PMA submissions, Panel-Track PMA supplement submissions, expedited original PMA submissions, 180-day supplement submissions, and premarket report submissions, issuance of one of the following letters is considered to be an FDA decision:
1. approval
2. approvable
3. approvable pending GMP inspection
4. not approvable
5. denial
B. For 510(k) submissions, issuance of one of the following letters is considered to be an FDA decision:
1. substantially equivalent (SE)
2. not substantially equivalent (NSE)
C. Submission of an unsolicited major amendment to an original PMA submission, Panel-Track PMA supplement submission, expedited original PMA submission, 180-day supplement submission, or premarket report submission extends the FDA decision goal date by the number of days equal to 75 percent of the difference between the filing date and the date of receipt of the amendment. The submission of the unsolicited major amendment is also considered an action that satisfies the first or later action goal, as applicable.
D. For BLA (original, efficacy supplement, or manufacturing supplement) submissions, the term "review and act on" is understood to mean the issuance of a complete action letter after the complete review of a filed complete application. The action letter, if it is not an approval, will set forth in detail the specific deficiencies and, where appropriate, the actions necessary to place the application in condition for approval.
E. For original BLA and BLA efficacy supplement resubmissions:
1. "Class 1" resubmitted applications are applications resubmitted after a complete response letter that include the following items only (or combinations of these items):
(a) Final printed labeling
(b) Draft labeling
(c) Safety updates submitted in the same format, including tabulations, as the original safety submission with new data and changes highlighted (except when large amounts of new information including important new adverse experiences not previously reported with the product are presented in the resubmission)>
(d) Stability updates to support provisional or final dating periods
(e) Commitments to perform Phase 4 studies, including proposals for such studies
(f) Assay validation data
(g) Final release testing on the last 1-2 lots used to support approval
(h) A minor reanalysis of data previously submitted to the application (determined by the Agency as fitting the "Class 1" category)
(i) Other minor clarifying information (determined by the Agency as fitting the "Class 1" category)
(j) Other specific items may be added later as the Agency gains experience with the scheme and will be communicated via guidance documents to industry.
2. "Class 2" resubmissions are resubmissions that include any other items, including any item that would require presentation to an advisory committee.
Different types of performance goals require different types of performance measures. FDA measures its success in meeting MDUFMA goals and commitments in two ways: using quantitative measures and using descriptive measures, depending on how the objective for a particular performance goal is described in FDA’s Commitment Letter. If the commitment letter provides an objective standard against which to measure FDA’s progress, quantitative measures are used. If the commitment letter does not provide an objective standard, FDA uses descriptive measures.
Quantitative progress is measured and described primarily through standard, quantifiable statistics (for example, number of submissions, mean performance, median performance, percent meeting a review time standard). Each quantitative goal has the following characteristics:
MDUFMA’s review performance goal progress is measured using quantitative methods.[9] Most of these goals use measures of success that become significantly more challenging over time. This approach recognizes that FDA must first hire and train new staff and rebuild review program infrastructures before it will be possible to make substantial progress in improving overall review performance, while providing interim goals that allow periodic evaluation of FDA’s progress towards the ultimate goals of the program.
Example: An example of where a performance goal is evaluated through quantitative measures is an expedited PMA, received during FY 2005, when FDA’s first action is a "major deficiency" letter. FDA will take that action (issue the letter) within 150 days of receipt of the expedited PMA [(FDA Commitment Letter, section I, paragraph B, Item 2(a)].
When quantitative measures cannot be used to evaluate FDA’s progress in implementing a performance goal, FDA uses descriptive measures to assess its performance. FDA reports its progress in narrative accounts that outline the specific actions FDA has taken; the results are attributed to those actions.
MDUFMA commitments use descriptive measures to assess performance (see Appendix A, section I, paragraphs I through P, of FDA’s Commitment Letter for detailed information). For descriptive measures, progress is reported through narrative accounts outlining specific actions taken, in addition to any results attributed to those actions. Descriptive measures:
FDA regards all of MDUFMA’s descriptive performance commitments to be in effect beginning with FY 2003 and will report progress towards achieving these commitments each year in the annual performance report.
Example: An example of where a performance goal is evaluated using descriptive measures is when FDA issues guidance on modular reviews under section 515(c)(3), and works with stakeholders to develop appropriate performance goals for the modular review program (FDA Commitment Letter, section I, paragraph L).
FDA measures its performance against applications in a receipt cohort. This methodology records performance on a submission in the statistics for the year it was received, regardless of when FDA ultimately acted on, approved, or cleared that submission. A consequence of this approach is that the statistics shown for a particular year may change from one report to the next. This is because, as time passes, FDA completes all work on more and more submissions. As more submissions are completed, the statistics for that year of receipt must be adjusted to reflect the new completions.
The performance goals of MDUFMA do not apply to device submissions received prior to FY 2003. Although FDA will work diligently to improve review performance for all applications, regardless of when they were received, submissions received prior to FY 2003 will not be reflected in the performance statistics used to evaluate FDA’s progress towards meeting MDUFMA goals. Submissions received since the start of FY 2003 (October 1, 2002) are subject to MDUFMA performance goals, and will be reflected in FDA’s performance statistics.
This table summarizes all of MDUFMA's quantifiable review performance goals (section I, goals A through H, in HHS Secretary Thompson's November 14, 2002, Commitment Letter).
| Activity | Review Time | Performance Level (by FY) (— indicates no quantitative goal) |
||||
|---|---|---|---|---|---|---|
| 2003 | 2004 | 2005 | 2006 | 2007 | ||
| PMAs, Panel-Track Supplements, Premarket Reports | ||||||
| FDA decision (approval, approvable, approvable pending GMP inspection, not approvable, denial) | 320 days | — | — | — | 80% | 90% |
| FDA decision (approval, approvable, approvable pending GMP inspection, not approvable, denial) | 180 days | — | — | — | — | 50% |
| First action – "major deficiency" letter | 150 days | — | — | 75% | 80% | 90% |
| First action – all other first actions (approval, approvable, approvable pending GMP inspection, not approvable, or denial) | 180 days | — | — | 75% | 80% | 90% |
| Second or later action – "major deficiency" letter | 120 days | — | — | 75% | 80% | 90% |
| Action on an amendment containing a complete response to a "major deficiency" or "not approvable" letter | 180 days | — | — | 75% | 80% | 90% |
| Action on an amendment containing a complete response to an "approvable" letter | 30 days | 90% | 90% | 90% | 90% | 90% |
| Expedited PMAs These goals apply when FDA has granted expedited status; the applicant has attended a pre-filing meeting; manufacturing facilities are ready for inspection; and the PMA is substantively complete as defined at the pre-filing meeting. |
||||||
| FDA decision (approval, approvable, approvable pending GMP inspection, not approvable, denial) | 300 days | — | — | 70% | 80% | 90% |
| First action – "major deficiency" letter | 120 days | — | — | 70% | 80% | 90% |
| First action – all other first actions (approval, approvable, approvable pending GMP inspection, not approvable, or denial) | 170 days | — | — | 70% | 80% | 90% |
| Second or later action – "major deficiency" letter | 100 days | — | — | 70% | 80% | 90% |
| Action on an amendment containing a complete response to a "major deficiency" or "not approvable" letter | 170 days | — | — | 70% | 80% | 90% |
| Action on an amendment containing a complete response to an "approvable" letter | 30 days | 90% | 90% | 90% | 90% | 90% |
| 180-day PMA Supplements | ||||||
| FDA decision (approval, approvable, approvable pending GMP inspection, not approvable, denial) | 180 days | — | — | 80% | 80% | 90% |
| First action – "not approvable" letter | 120 days | — | — | 80% | 85% | 90% |
| First action – all other first actions (approval, approvable, approvable pending GMP inspection, or denial) | 180 days | — | — | 80% | 85% | 90% |
| Action on an amendment containing a complete response to a "not approvable" letter | 160 days | — | — | 80% | 85% | 90% |
| 510(k)s | ||||||
| FDA decision (SE/NSE) | 90 days | — | — | 75% | 75% | 80% |
| First action – "additional information" letter | 75 days | — | — | 70% | 80% | 90% |
| Second or later action | 60 days | — | — | 70% | 80% | 90% |
| Biologics Licensing Applications - BLAs | ||||||
| Review and act on standard original BLAs (issue "complete action" letter) | 10.0 months | — | — | — | 75% | 90% |
| Review and act on priority original BLA submissions (issue "complete action" letter) | 6.0 months | — | — | — | 75% | 90% |
| BLA Supplements | ||||||
| Review and act on standard BLA efficacy supplements (issue "complete action" letter) | 10.0 months | — | — | — | 75% | 90% |
| Review and act on priority BLA efficacy supplements (issue "complete action" letter) | 6.0 months | — | — | — | 75% | 90% |
| Review and act on BLA manufacturing supplements that require prior approval (issue "complete action" letter) | 4.0 months | — | — | — | 75% | 90% |
| BLA Resubmissions, BLA Supplement Resubmissions | ||||||
| Review and act on a "Class 1" resubmission to an original BLA or BLA efficacy supplement (issue "complete action" letter) | 2.0 months | — | — | 75% | 80% | 90% |
| Review and act on a "Class 2" resubmission to an original BLA or BLA efficacy supplement (issue "complete action" letter) | 6.0 months | — | — | 75% | 80% | 90% |
Note: Definitions for the terms used here are provided in Section III of the FDA's Commitment Letter
1. Section 738(g) of FD&C Act, as amended by MDUFMA. Except where noted, all statutory citations in this report are to the FD&C Act, as amended by MDUFMA.
2. HHS Secretary submitted the required letter to Congress on November 14, 2002 (Congressional Record, November 19, 2002, p. S11549). For convenience, this report refers to this letter as “FDA’s Commitment Letter.” The complete text of the letter is provided in Appendix A.
3. All submissions under MDUFMA are measured by the cohort year of original submission. Unless all submissions in a cohort are completed, only a preliminary performance assessment can be provided for that cohort.
4. FDA did not receive any premarket reports in FY 2003 through FY 2007.
5. The MDUFMA Cohort for 510(k)s excludes submissions that were closed for any reason other than an SE or NSE decision (for example, when FDA finds that a 510(k) was not required). Each MDUFMA cohort number is subject to change until that cohort is closed.
6. Bundling refers to the inclusion of multiple devices or multiple indications for use for a device in a single premarket submission, including products subject to the device and biologics license application (BLA) authorities, for purposes of review and user fee payment.
7. FY 2006 number was updated to reflect corrections to the electronic radiation safety reports presented in the FY 2006 MDUFMA Performance Report.
8. This text was edited from the original version. “Not approvable” was taken out of the list of “All other first action letters.” Because “Not approvable” letter is already captured under the “First Action” goal of 120 days, it should not be repeated under the “All other first actions” goal of 180 days.
9. These quantitative goals are defined in section I, paragraphs A through H, of FDA’s Commitment Letter. A tabular summary of all of MDUFMA’s objective performance goals is provided in Attachment C. An example of a quantitative goal is for Expedited PMAs: “70 percent of submissions received in fiscal year 2005 will have an FDA decision in 300 days.” This is a quantitative goal because it applies to a defined category of applications (expedited PMAs), involves a defined type of action (an FDA decision), sets an objective review time standard (300 days), has a quantifiable measure of successful performance (70 percent of submissions), and applies within a specific time frame (FY 2005) (see section I, paragraph B, goal 3(a) of FDA’s Commitment Letter in Appendix A).
 Department of Health and Human Services
Department of Health and Human Services 
Food and Drug Administration
| This report was prepared by FDA's Office of Planning in collaboration with the Center for Biologics Evaluation and Research (CBER) and the Center for Devices and Radiological Health (CDRH). For information on obtaining additional copies contact: Office of Planning (HFP-10) Food and Drug Administration 5600 Fishers Lane Rockville, Maryland 20857 Phone: 301-827-5292 FAX: 301-827-5260 This report is available on the FDA Home Page at http://www.fda.gov. |
![]()