


CBER Presentation
Printable Version
Note: Documents in PDF format require the Adobe Acrobat Reader®. If you experience problems with PDF documents, please download the latest version of the Reader®.
IND "202" Clinical Perspective
ISCT 6th Annual Somatic Cell Therapy Symposium
9/25-27/2006
Steven Hirschfeld, MD PhD
Office of Cellular, Tissue and Gene Therapies
Center for Biologics Evaluation and Research
Food and Drug Administration
Topics Covered
- IND activity for somatic cell therapies in CBER OCTGT
- Pre-IND meeting
- IND Processing
- Document preparation
- Review Team Assignments
- Review Process
- Clinical Review
- Priorities
- Common Clinical Hold Issues
- IND Annual Report Review
What is an IND?
- Investigational New Drug application
- Mechanism to allow interstate transport of investigational agents for clinical research
- Provide patient protection through Federal oversight
- Authority for IND mechanism comes from Food, Drug & Cosmetic Act Section 505
- IND regulations may be found in the Code of Federal Regulations (CFR) Title 21 Part 312
21CFR312.22
- (a) FDA's primary objectives in reviewing an IND are, in all phases of the investigation, to assure the safety and rights of subjects, and, in Phase 2 and 3, to help assure that the quality of the scientific evaluation of drugs is adequate to permit an evaluation of the drug's effectiveness and safety.
Overview of Therapeutic Development
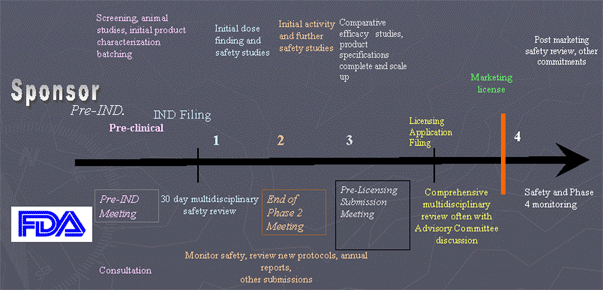
Pre IND Meeting
- Non-binding advice provided as courtesy
- Request is made to FDA.
- Meeting is scheduled.
- At least 30 days prior to scheduled meeting potential IND applicant submits to FDA a meeting package consisting of
- background information
- list of questions grouped by type (Product, Preclinical, Clinical and Regulatory)
- FDA review team with members from each discipline will review package and hold an internal meeting to responses to the submitted questions
- Draft responses to the questions are usually communicated to the potential applicant prior to the scheduled meeting
- Potential applicant has option to
- cancel meeting if draft answers are satisfactory or
- hold meeting but focus only on selected issues or
- hold meeting and discuss all questions and responses
IND Processing
- IND Package arrives at FDA and is processed
- Following processing, review assignments are made for relevant disciplines. Usual are product (CMC), preclinical pharmacology/toxicology, and clinical. As needed a statistical review is requested
- Legal requirement for 30 day review based on calendar (no holidays allowed!) beginning when the submission is logged into document control
- FDA internal reviews are based on standardized templates
- Clarifications from the IND sponsor are usually sought during the review process with the goal of avoiding clinical hold as noted in 21CFR312.42(c)
Clinical Hold Criteria
- Risks are unreasonable and significant
- Investigators not qualified
- Investigator brochure false or misleading
- Insufficient information to assess risk
- Gender exclusion for a condition that occurs in both men and women
Clinical Hold criteria are described in 21CFR312.42
IND Review Completion
- Approximately one week prior to the due date the review team makes a preliminary determination if a clinical hold may be necessary
- If no clinical hold is issued, sponsor may begin clinical investigation after 30th day with IRB approval.
- No formal communication from FDA is necessary to activate an IND.
Clinical Hold Issuance
- If deficiencies cannot be resolved FDA will issue a clinical hold
- IND applicant is informed by telephone prior to or on Day 30 following IND submission
- A letter from the FDA is sent to the IND applicant within 30 days of the telephone call
- listing the clinical hold issues
- Stating what is expected to remove the clinical hold
- Sponsor may reply at any time following the telephone call with a Complete Response to Clinical Hold
Clinical Hold
- A clinical hold may be issued at any time
- For ongoing studies usually adverse events trigger the clinical hold
- If only some and not all studies under an active IND are placed on clinical hold, the IND is under Partial Hold
- If all investigations under an IND remain on clinical hold for more than one year, the IND is automatically converted to inactive status
Clinical Review Elements
- All elements as outlined in 21CFR 312.23 are included in IND submission
- Adequate rationale and preclinical data to justify starting dose and schedule
- Appropriate patient population
- Acceptable dose escalation schema
- Staggered enrollment for new agents
- Adequate safety monitoring based on patient population and anticipated toxicities
IND Requirements based on 21CFR312.23
- Cover sheet of a Form 1571 filled out by the sponsor. Each Associate investigator must have an additional Form designated as 1572. Both forms are available at http://forms.psc.gov/forms/FDA/fda.html
- Table of contents
- Introductory statement and summary of general investigational plan
- Product information including chemistry, manufacturing and control information
- Pharmacology and toxicology information
- Summary of prior human experience with the product, if applicable.
- Clinical protocols. Specific listings and requirements are noted in the regulations for various protocol types.
- Special topic information such as dependence or abuse potential, radioactive information, and plans for pediatric studies .
Clinical Review Elements
- Adverse event reporting consistent with 21CFR312.32
- Patient withdrawal criteria
- Study stopping criteria
- Adequate informed consent procedure
- Study conduct monitoring in compliance with Good Clinical Practice (ICH E6)
Investigator Brochures
- An Investigator's Brochure is required
- if the planned investigation will occur at multiple sites and
- the sponsor is not a sponsor-investigator.
- Most academic investigator initiated studies that file for an IND are considered sponsor-investigator studies and will not require an Investigator's Brochure unless the product is supplied by a third party.
- The IND regulations refer to the requirements for Investigator Brochure in two complementary sections and both sections apply
- 21CFR312.23 (a) (5)
- 21CFR312.55.
Common Clinical Reasons for Clinical Hold
Patient population:
- Eligibility and/or exclusion criteria inappropriate
- Number of subjects not specified or unreasonable
Starting dose:
- Insufficient data to support the intended starting dose
- Product preparation or formulation inadequately described
Dose regimen:
- Administration of product risky or inadequately described
- Proposed dose increases too aggressive
- Failure to stagger enrollment of new product with unknown risks
- Dose modification plan unreasonable
- Repeat treatment plan unreasonable or not supported
Safety monitoring:
- Anticipated toxicities inadequately monitored
- Lack of appropriate Toxicity Scale
- Individual Patient Treatment Discontinuation Criteria absent or unreasonable
- Study Stopping Rules absent or unreasonable
- Withdrawn subjects not adequately followed
- Long term follow up for patients absent or inadequately described
- Adverse event reporting procedures inadequate