


CBER Presentation
PDF Version (102)
Note: Documents in PDF format require the Adobe Acrobat Reader®. If you experience problems with PDF documents, please download the latest version of the Reader®.
Facility Design and CGMP Considerations for Cell Therapy Products
ISCT- 6th Annual Symposium
September 25-27, 2006
James Crim
HHS/FDA/CBER/OCBQ/DMPQ
SCOPE
- Regulatory Overview
- Current Good Manufacturing Practices (cGMPs) - Expectations During Development
- Basic facility considerations
- Facilities/Personnel
- Environmental Monitoring
- Contamination and cross contamination control
- Aseptic Processing
Regulatory Considerations
- IND regulations (21 CFR 312) - patient safety and clinical trials
- Biologics regulations (21 CFR 600s) - licensing requirements
- CGMPs (21 CFR 211s) - current good manufacturing practices for drugs/biologics
- Combination products (21 CFR 800s)
CGMP Continuum: Expectations During Development
- GMPs expected throughout clinical studies, though level of control and validation will vary on the process and critical nature of the issue
- Documented control over the facility, equipment, and process
- Expect control to increase as product moves from one phase to the next
Consider some of the following elements of CGMP:
- Facility design to control operations
- Adequate documentation/records
- Production and process controls
- Quality control/assurance
- Validation/process control
- Equipment calibrated/qualified
- Personnel training and certification
- Environmental monitoring
Facility Design
- Manufacturing areas designed for aseptic processing - smooth, easily cleaned surfaces, etc.
- Designed to control the manufacturing environment (personnel and process)
- Adequate and separate areas, for various activities (receipt of materials, testing, manufacturing)
- HEPA-filtered air in manufacturing areas; higher control (classification) for critical manufacturing steps
- Product type and makeup
- Stage of manufacturing
- Scale of manufacturing
- Material and personnel flows designed to maximize efficiency and minimize product mix-ups
- Concurrent vs. campaigning - impact on HVAC, cleaning and personnel
Facility Design considerations: Product Issues
- Will the entire manufacturing process be performed in the facility?
- Nature of the starting material - cell culture vs. recombinant cell line
- Nature of the process - open vs. closed systems; fermentation/cell culture, purification, etc.
- Multi-product or patient manufacturing?
- Facility intended to be licensed or limited to IND products?
Facility Design: Multi-Product Issues
- Campaign vs. concurrent production will impact on design and operation of the facility
- Commercial vs. investigational product manufacturing
- Dedicated vs. shared equipment
- Multiple patient cells
Facilities/Personnel
- Personnel practice universal precautions when processing biological materials such as cells or tissues
- Unidirectional flow of personnel and processed material
- Temporal segregation of processing activities, if possible
- Gowning program designed to protect the product from contamination and keep airborne particulates away from the product and prevent the transfer of particulates from one manufacturing environment to another environment of higher classification
Environmental Monitoring
- Purpose
- To demonstrate that environment quality is consistently within specified levels.
- To provide a timely and sensitive warning if the environmental quality or its control is becoming or have become unacceptable.
- To initiate a timely, comprehensive planed course of action whenever environmental monitoring results are indicative of unacceptable environmental quality or control (i.e., "excursion" or OOS).
- How much environmental monitoring is needed and when?
- The main goal is to protect the product by demonstrating that the class 100 environment has been maintained.
- Should have qualified Biosafety Cabinets of appropriate air classification and on a maintenance plan for filter testing.
- Should routinely monitor (if even settling plate) during processing of patient cells or tissues to ensure no additional microbial contamination.
- Should be working towards process simulations to ensure aseptic processing.
- Operators should be trained and practice due diligence with regard to aseptic manipulations.
Routine dynamic monitoring of the "clean" environment and operations is important to insure that modes of bioburden introduction are under control. The recommendation is to at least monitor viable particulates during aseptic processing and understand the airflow in the hood and pressure differentials in areas of operations as the pressure differentials may provide an indication that the area is suitable for use.
Examples of operations that may be performed in classified areas
| Class/(ISO equivalent) | Example of operation |
|---|---|
| Class 100/ ISO 5 |
|
| Class 1000/ ISO 6 Class 10,000/ISO 7 |
|
| Class 100,000/ ISO 8* |
|
* If additional microbiological controls are required the procedure should be performed in a Class 10,000 area
An example of a floor plan
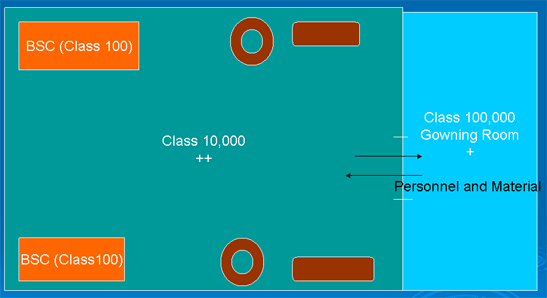
Controls for preventing contamination and cross contamination:
- Campaign manufacturing and area/equipment clearance
- Labeling system
- Segregation and Tracking of Patient Material
- Use of Sterile Transfer Connecting Devices
- Cleaning and Sanitization
- Flow of Product and Waste Material
- Gowning Program
- Preventing microbial contamination (Aseptic Processing)
Aseptic Processing
- What is aseptic processing (technique)?
- Where does aseptic processing start?
The ability of personnel to manipulate sterile preparations, sterile packaging components, and sterile administration devices in a way that excludes theintroduction of viable microorganisms.
It may depend on the process, it may have a sterilizing step prior to filling or some products can not be sterilized so aseptic processing occurs from beginning to the end.
Critical input elements to successful aseptic processing:
- Personnel performance (gowning and technique)
- Environmental quality and control
- Validated, controlled sterilization of all product and added ingredients, container/closures, equipment, utensils, and product-contract surfaces
- Media Challenge - worse case scenarios, routine dynamic environmental monitoring, equipment and environmental capabilities, sterility needs for supporting process stream contact materials
In Summary..
- FDA recognizes changing nature of clinical studies; need for sliding scale approach to meeting cGMPs
- Key production steps, equip. and facilities need to be under documented control
- Patient safety cannot be compromised
- Testing alone does not assure a quality product
- QC/QA needed at early stages
- Suggest facility be designed to accommodate current and future needs
- Suggest facility be designed to be as flexible as possible
- Suggest that the facility be designed, operated and controlled to the highest level possible (room classifications, pressure differentials, monitoring, etc.)
Special thanks to.
Jay Eltermann - OCBQ/DMPQ
Laurie Norwood - OCBQ/DMPQ
For more information..
- Reviews/meetings with DMPQ:
(301) 827-3031, (301) 827-3536 fax - Technical issues: webmaster@cber.fda.gov
- Manufacturers assistance: matt@cber.fda.gov
- CBER SOPP for meetings: http://www.fda.gov/cber/regsopp/81011.htm