

|
|
(Posted: 12/17/2001)
How to Find a Safety-Related Labeling ChangeUse the drop-down menu to select a product by brand name. |
MedWatch
Home | What's New | About
Medwatch | How to Report | Submit
Report | Safety Info
Continuing Education | Download
PDF | Comments | Privacy
Statement
ATACAND (candesartan cilexetil) Tablets,
ATACAND HCT (candesartan cilexetil/hydrochlorothiazide) Tablets
[November 28, 2001: AstraZeneca]
ADVERSE REACTIONS
Post-Marketing Experience
Revised for ATACAND Tablets and added as a new subsection to the ATACAND HCT Tablets.
Other adverse events reported for candesartan cilexetil where a casual relationship could not be established include very rare cases of neutropenia, leukopenia and agranulocytosis.
The following have been very rarely reported in post-marketing experience:
Digestive: Abnormal hepatic function and hepatitis.
Hematologic: Neutropenia, leukopenia, and agranulocytosis.
Skin and Appendages Disorders: Pruritus and urticaria
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
BRANCHAMIN 4% Amino Acids Injectable
[November 14, 2001: Baxter Healthcare]
WARNINGS
Added
Hyperammonemia has been reported to occur in infants receiving various amino acid supplementation regimens. Blood ammonia monitoring may be prudent in 4% BranchAmin infusions.
PRECAUTIONS
Pediatric Use
Revised
Safety and effectiveness in children have not been established.
The safety and effectiveness of 4% BranchAmin (Branched Chain Amino Acid) Injection in pediatric patients have not been established by adequate and well-controlled trials.
DOSAGE AND ADMINISTRATION
Revised
Do not administer unless solution is clear.
Do not administer unless solution is clear and seal is intact.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
CALCIJEX (calcitriol) Injection
[November 16, 2001: Abbott Laboratories]
PRECAUTIONS:
Pediatric Use
Revised
Safety and efficacy of Calcijex in pediatric patients have not been established.
"The safety and effectiveness of Calcijex were examined in a 12-week randomized, double-blind, placebo-controlled study of 35 pediatric patients, aged 13-18 years, with end-stage renal disease on hemodialysis. Sixty-six percent of the patients were male, 57% were African-American, and nearly all had received some form of vitamin D therapy prior to the study. The initial dose of Calcijex was 0.5 mcg, 1.0 mcg, or 1.5 mcg, 3 times per week, based on baseline a iPTH level of less than 500 pg/ml, 500-1000 pg/ml, or greater than 1000 pg/ml, respectively. The dose of Calcijex was adjusted in 0.25 mcg increments based on the levels of serum iPTH, calcium, and Ca x P. The mean baseline levels of iPTH were 769 pg/ml for the 16 Calcijex-treated patients and 897 pg/ml for the 19 placebo-treated subjects. The mean weekly dose of Calcijex ranged from 1.0 mcg to 1.4 mcg. In the primary efficacy analysis, 7 of 16 (44%) subjects in the Calcijex group had 2 consecutive 30% decreases from baseline iPTH compared with 3 of 19 (16%) patients in the placebo group (95% CI for the difference between groups -6%, 62%). One Calcijex- treated patient experienced transient hypercalcemia (> 11.0 mg/dl), while 6 of 16 (38%) Calcijex-treated patients vs. 2 of 19 (11%) placebo-treated patients experienced Ca x P >75."
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
COREG (carvedilol) Tablets
[November 1, 2001: GlaxoSmithkKine]
CLINICAL PHARMACOLOGY
Pharnacodynamics and Clinical trials
Congestive Heart Failure
Pharmacodynamics
4th paragraph revised
Among 839 patients with NYHA class II-III heart failure treated for 26 to 52 weeks in 4 U.S. placebo-controlled trials, the average left ventricular ejection fraction (EF) measured by radionuclide ventriculography increased by 8 9 EF units (%) in Coreg patients and by 2 EF units in placebo patients ( between group difference of 6 EF units) This treatment effect was. at a target dose of 25-50 mg b.i.d. The effects of carvedilol on ejection fraction were related to dose. Doses of 6.25 mg b.i.d., 12.5 mg b.i.d. and 25 mg b.i.d. were associated with placebo-corrected increases in EF of 5 EF units, 6 EF units and 8 EF units, respectively; each of these effects were nominally statistically significant in each trial.
CLINICAL TRIALS
Congestive Heart Failure
revised
A total of 3946 patients with mild to severe heart failure were evaluated in placebo–controlled studies of carvedilol.
In the largest study (COPERNICUS), 2289 patients with heart failure at rest or with minimal exertion and left ventricular ejection fraction <25% (mean 20%), despite digitalis (66%), diuretics (99%), and ACE inhibitors (89%) were randomized to placebo or carvedilol. Carvedilol was titrated from a starting dose of 3.125 mg twice daily to the maximum tolerated dose or up to 25 mg twice daily over a minimum of 6 weeks. Most subjects achieved the target dose of 25 mg. The study was conducted in Eastern and Western Europe, the United States, Israel, and Canada. Similar numbers of subjects per group (about 100) withdrew during the titration period.
The primary end point of the trial was all-cause mortality, but cause-specific mortality and the risk of death or hospitalization (total, cardiovascular [CV] or congestive heart failure [CHF]) were also examined. The developing trial data were followed by a data monitoring committee, and mortality analyses were adjusted for these multiple looks. The trial was stopped after a median follow-up of 10 months because of an observed 35% reduction in mortality (from 19.7% per patient year on placebo to 12.8% on carvedilol, hazard ratio 0.65, 95% CI 0.52 – 0.81, p=0.0014, adjusted). The results of COPERNICUS are shown in Table 1.
Table 1. Results of COPERNICUS
|
End point |
Placebo N = 1133 |
Carvedilol N=1156 |
Hazard ratio (95% CI) |
% Reduction |
NominalP value |
|
|
|
|
|||
|
Mortality |
190 |
130 |
0.65 0.52 – 0.81 |
35 |
0.00013 |
|
Mortality + all hospitalization |
507 |
425 |
0.76 0.67 – 0.87 |
24 |
0.00004 |
|
Mortality + CV hospitalization |
395 |
314 |
0.73 0.63 – 0.84 |
27 |
0.00002 |
|
Mortality + CHF hospitalization |
357 |
271 |
0.69 0.59 – 0.81 |
31 |
0.000004 |
Figure 1. Survival analysis for COPERNICUS (intent-to-treat)

Effects were similar in patients with and without diabetes based on data from both COPERNICUS and the U.S. trials described below.
The effect on mortality was principally the result of a reduction in the rate of sudden death among patients without worsening heart failure.
Patients' global assessments showed significant improvement following treatment with carvedilol in COPERNICUS.
The protocol also specified that hospitalizations would be assessed. Fewer patients on Coreg than on placebo were hospitalized for any reason (198 vs. 268, p=0.0001), for cardiovascular reasons (246 vs. 314, p=0.0003), or for worsening heart failure (372 vs. 432, p=0.0029).
Coreg had a consistent and beneficial effect on all-cause mortality as well as the combined endpoints of all-cause mortality plus hospitalization (total, CV or for heart failure) in the overall study population and in all subgroups examined, including men and women, elderly and non-elderly, black and non-black.
Carevedilol was also studied in five other multi-center, placebo-controlled studies.
Four U.S. multicenter, double-blind, placebo-controlled studies enrolled 1094 patients (696 randomized to carvedilol) with NYHA class II-III heart failure and ejection fraction <0.35. The vast majority were on digitalis, diuretics, and an ACE inhibitor at study entry. Patients were assigned to the studies based upon exercise ability. An Australia-New Zealand double-blind, placebo-controlled study enrolled 415 patients (half randomized to carvedilol) with less severe heart failure. All protocols excluded patients expected to undergo cardiac surgery during the 6 to 12 months of double-blind follow-up transplantation during the 7.5 to 15 months of double-blind follow-up.. All randomized patients had tolerated a 2-week course on carvedilol 6.25 mg b.i.d.
Mortality: Mortality was not a planned end-point in any of the U.S. or Austrailia-New Zealand studies. Overall, in the U.S. trials, mortality was reduced, nominally significantly so in 2 studies, but the actual effect size and statistical significance of this observation are difficult to define.
Mortality: Overall, in these four U.S. trials, mortality was reduced, nominally significantly so in 2 studies.
INDICATIONS AND USAGE
Congestive Heart Failure
first paragraph revised
Coreg is indicated for the treatment of mild or moderate (NYHA class II or III) severe heart failure of ischemic or car-diomyopathic origin, in conjunction with usually in addition to diuretics, ACE inhibitor and digitalis, diuretics, and ACE inhibitor, to reduce the progression of disease as evidenced by cardiovascular death, cardiovascular hospitalization, or the need to adjust other heart failure medications. to increase survival and, also, to reduce the risk of hospitalization.
CONTRAINDICATIONS
first paragraph revised
Coreg is contraindicated in patients with NYHA class IV decompensated cardiac failure requiring intravenous inotropic therapy, bronchial asthma (two cases of death from status asthmaticus have been reported in patients receiving single doses of Coreg ) or related bronchospastic conditions, second- or third-degree AV block, sick sinus syndrome or severe bradycardia (unless a permanent pacemaker is in place) ); or in patients with cardiogenic shock or severe bradycardia. who have decompensated heart failure requiring the use of intravenous inotropic therapy. Such patients should first be weaned from intravenous therapy before initiating Coreg.
WARNINGS
Hepatic Injury
2nd and 3rd paragraphs revised
In controlled studies of primarily mild-to-moderate congestive heart failure, the incidence of liver function abnormalities reported as adverse experiences was 5.0% (38 of 765 patients) in patients receiving Coreg and 4.6% (20 of 437 patients) in those receiving placebo. Three patients receiving carvedilol Coreg (0.4%) and two patients receiving placebo (0.5%) in placebo-controlled trials withdrew for abnormal hepatic function. Similarly, in a long-term, placebo-controlled trial in severe heart failure, there was no difference in the incidence of liver function abnormalities reported as adverse experiences between patients receiving Coreg and those receiving placebo. No patients receiving Coreg and one patient receiving placebo (0.09%) withdrew for hepatitis. In addition, patients treated with Coreg had lower values for hepatic transaminases than patients treated with placebo, possibly because Coreg-induced improvements in cardiac function led to less hepatic congestion and/or improved hepatic blood flow.
Hepatic injury has been reversible and has occurred after short- and/or long-term therapy with minimal clinical symptomatology. No deaths due to liver function abnormalities have been reported in association with the use of Coreg.
Diabetes and Hypoglycemia:
first sentence revised
In general, b -blockers may mask some of the manifestations of hypoglycemia, particularly tachycardia.
PRECAUTIONS
General
first paragraph revised
Since Coreg (carvedilol) has b -blocking activity, it should not be discontinued abruptly, particularly in patients with ischemic heart disease. Instead, it should be discontinued over 1 to 2 weeks, whenever possible.
3rd paragraph revised
In clinical trials of primarily mild-to-moderate heart failure, Hhypotension and postural
hypotension occurred in 9.7% and syncope in 3.4% of patients receiving carvedilolCoreg compared to 3.6% and 2.5% of placebo patients, respectively. The risk for these events was highest during the first 30 days of dosing, corresponding to the up-titration period and was a cause for discontinuation of therapy in 0.7% of carvedilolCoreg patients, compared to 0.4% of placebo patients. In a long-term, placebo-controlled trial in severe heart failure (COPERNICUS), hypotension and postural hypotension occurred in 15.1% and syncope in 2.9% of heart failure patients receiving Coreg compared to 8.7% and 2.3% of placebo patients, respectively. These events were a cause for discontinuation of therapy in 1.1% of Coreg patients, compared to 0.8% of placebo patients.
7th paragraph revised
Worsening cardiac heart failure or fluid retention may occur during up-titration of carvedilol. If such symptoms occur, diuretics should be increased and the carvedilol dose should not be advanced until clinical stability resumes (see DOSAGE AND ADMINISTRATION). Occasionally, it is necessary to lower the carvedilol dose or temporarily discontinue it. Such episodes do not preclude subsequent successful titration of or a favorable response to carvedilol. In a placebo-controlled trial of patients with severe heart failure, worsening heart failure during the first 3 months was reported to a similar degree with carvedilol and with placebo. When treatment was maintained beyond 3 months, worsening heart failure was reported less frequently in patients treated with carvedilol than with placebo. Worsening heart failure observed during long-term therapy is more likely to be related to the patients’ underlying disease than to treatment with carvedilol.
Deleted
Hypertensive Patients with Left Ventricular Failure: In hypertensive patients who have congestive heart failure controlled with digitalis, diuretics and/or an angiotensin-converting enzyme inhibitor, Coreg (carvedilol) may be used. However, since it is likely that such patients are dependent, in part, on sympathetic stimulation for circulatory support, it is recommended that dosing follow the instructions for patients with congestive heart failure.
Geriatric Use
first paragraph revised
Of the 765 patients with congestive heart failure randomized to Coreg in U.S. clinical trials, 31% (235) were 65 years of age or older. Of 1,869 patients receiving Coreg in congestive heart failure trials worldwide, 39% were 65 years of age or older. Of the 1156 patients randomized to Coreg in a long-term, placebo-controlled trial in severe heart failure, 47% (547) were 65 years of age or older. Of 3,025 patients receiving Coreg in congestive heart failure trials worldwide, 42% were 65 years of age or older. There were no notable differences in efficacy or the incidence of adverse events between older and younger patients.
ADVERSE REACTIONS
Congestive Heart Failure
revised
Coreg has been evaluated for safety in congestive heart failure in more than 1,900 3,000 patients worldwide of whom 1,300 more than 2100 participated in U.S. placebo-controlled clinical trials. Approximately 54% 60%of the total treated population received Coreg for at least 6 months and 20% 30% received Coreg for at least 12 months. The adverse experience profile of Coreg in congestive heart failure patients was consistent with the pharmacology of the drug and the health status of the patients. In U.S. clinical trials comparing Coreg in in mild-to-moderate heart failure that compared Coreg daily doses up to 100 mg (n=765) to placebo (n=437), 5.4% of Coreg patients discontinued for adverse experiences vs. 8.0% of placebo patients. In a multinational clinical trial in severe heart failure (COPERNICUS) that compared Coreg in daily doses up to 50 mg (n=1156) with placebo (n=1133), 9.4% of Coreg patients discontinued treatment for adverse experiences vs. 11.2% of placebo patients.
Table 1 2 shows adverse events reported in patients with mild-to-moderate heart failure enrolled in U.S. placebo-controlled clinical trials of congestive heart failure patients that occurred with an incidence of greater than 2% regardless of causality and were more frequent in drug-treated patients than placebo-treated patients. Median study medication exposure was 6.33 months for both Coreg (carvedilol) and placebo patients. Shown are adverse events that occurred more frequently in drug-treated patients than placebo-treated patients with an incidence >2% regardless of causality. Median study medication exposure was 6.33 months for both carvedilol and placebo patients.
|
Adverse Reactions |
Withdrawals |
|||
|
|
Placebo |
|
Placebo |
|
|
Autonomic Nervous System |
||||
|
Sweating increased |
2.9 |
2.1 |
-- |
-- |
|
Body as a Whole |
||||
|
Fatigue |
23.9 |
22.4 |
0.7 |
0.7 |
|
Chest pain |
14.4 |
14.2 |
0.1 |
-- |
|
Pain |
8.6 |
7.6 |
-- |
0.2 |
|
Injury |
5.9 |
5.5 |
-- |
-- |
|
Drug level increased |
5.1 |
3.7 |
-- |
0.2 |
|
Edema generalized |
5.1 |
2.5 |
-- |
-- |
|
Edema dependent |
3.7 |
1.8 |
-- |
-- |
|
Fever |
3.1 |
2.3 |
-- |
-- |
|
Edema legs |
2.2 |
0.2 |
0.1 |
0.2 |
|
Cardiovascular |
||||
|
Bradycardia |
8.8 |
0.9 |
0.8 |
-- |
|
Hypotension |
8.5 |
3.4 |
0.4 |
0.2 |
|
Syncope |
3.4 |
2.5 |
0.3 |
0.2 |
|
Hypertension |
2.9 |
2.5 |
0.1 |
-- |
|
AV block |
2.9 |
0.5 |
-- |
-- |
|
Angina pectoris aggravated |
2.0 |
1.1 |
-- |
-- |
|
Central Nervous System |
||||
|
Dizziness |
32.4 |
19.2 |
0.4 |
-- |
|
Headache |
8.1 |
7.1 |
0.3 |
-- |
|
Paresthesia |
2.0 |
1.8 |
0.1 |
-- |
|
Gastrointestinal |
||||
|
Diarrhea |
11.8 |
5.9 |
0.3 |
-- |
|
Nausea |
8.5 |
4.8 |
-- |
-- |
|
Abdominal Pain |
7.2 |
7.1 |
0.3 |
-- |
|
Vomiting |
6.3 |
4.3 |
0.1 |
-- |
|
Hematologic |
||||
|
Thrombocytopenia |
2.0 |
0.5 |
0.1 |
-- |
|
Metabolic |
||||
|
Hyperglycemia |
12.2 |
7.8 |
0.1 |
-- |
|
Weight increase |
9.7 |
6.9 |
0.1 |
0.5 |
|
Gout |
6.3 |
6.2 |
-- |
-- |
|
BUN increased |
6.0 |
4.6 |
0.3 |
0.2 |
|
NPN increased |
5.8 |
4.6 |
0.3 |
0.2 |
|
Hypercholesterolemia |
4.1 |
2.5 |
-- |
-- |
|
Dehydration |
2.1 |
1.6 |
-- |
-- |
|
Hypervolemia |
2.0 |
0.9 |
-- |
-- |
|
Musculoskeletal |
||||
|
Back pain |
6.9 |
6.6 |
-- |
-- |
|
Arthralgia |
6.4 |
4.8 |
0.1 |
0.2 |
|
Myalgia |
3.4 |
2.7 |
-- |
-- |
|
Resistance Mechanism |
||||
|
Upper respiratory tract infection |
18.3 |
17.6 |
-- |
-- |
|
Infection |
2.2 |
0.9 |
-- |
-- |
|
Respiratory |
||||
|
Sinusitis |
5.4 |
4.3 |
-- |
-- |
|
Bronchitis |
5.4 |
3.4 |
-- |
0.2 |
|
Pharyngitis |
3.1 |
2.7 |
-- |
-- |
|
Urinary/Renal |
||||
|
Urinary tract infection |
3.1 |
2.7 |
-- |
-- |
|
Hematuria |
2.9 |
2.1 |
-- |
-- |
|
Vision |
||||
|
Vision abnormal |
5.0 |
1.8 |
0.1 |
-- |
Incidence >2%, Regardless of Causality; Withdrawal Rates due to Adverse Events
In addition to the events in Table 1, asthenia, cardiac failure, flatulence, anorexia, dyspepsia, palpitation, extrasystoles, hyperkalemia, arthritis, angina pectoris, insomnia, depression, anemia, viral infection, dyspnea, coughing, respiratory disorder, rhinitis, rash, and leg cramps were also reported, but rates were equal to, or more common in, placebo-treated patients.
The following adverse events were reported more frequently with Coreg in U.S. placebo-controlled trials in patients with congestive heart failure:
Incidence >1% to <2%
Body as a Whole: Peripheral edema, allergy, sudden death, malaise, hypovolemia.
Cardiovascular: Fluid overload, postural hypotension.
Central and Peripheral Nervous System: Hypesthesia, vertigo.
Gastrointestinal: Melena, periodontitis.
Liver and Biliary System: SGPT increased, SGOT increased.
Metabolic and Nutritional: Hyperuricemia, hypoglycemia, hyponatremia, increased alkaline phosphatase, glycosuria.
Platelet, Bleeding and Clotting: Prothrombin decreased, purpura.
Psychiatric: Somnolence.
Reproductive, male: Impotence.
Urinary System: Abnormal renal function, albuminuria.
Table 2. Adverse Events (% Occurrence and % Withdrawals) Occurring More Frequently with Coreg Than with Placebo in Patients with Mild-to-Moderate Heart Failure Enrolled in U.S. Heart Failure Trials (Incidence >2%, Regardless of Causality)
|
Adverse Reactions |
Withdrawals |
||||
|
Coreg |
Placebo |
Coreg |
Placebo |
||
|
(n=765) |
(n=437) |
(n=765) |
(n=437) |
||
|
% occurrence |
% occurrence |
% withdrawals |
% withdrawals |
||
|
Autonomic Nervous System |
|||||
|
Sweating increased |
3 |
2 |
— |
— |
|
|
Body as a Whole |
|||||
|
Fatigue |
24 |
22 |
0.7 |
0.7 |
|
|
Pain |
9 |
8 |
— |
0.2 |
|
|
Digoxin level increased |
5 |
4 |
— |
0.2 |
|
|
Edema generalized |
5 |
3 |
— |
— |
|
|
Edema dependent |
4 |
2 |
— |
— |
|
|
Fever |
3 |
2 |
— |
— |
|
|
Edema legs |
2 |
-— |
0.1 |
0.2 |
|
|
Cardiovascular |
|||||
|
Bradycardia |
9 |
1 |
0.8 |
— |
|
|
Hypotension |
9 |
3 |
0.4 |
0.2 |
|
|
AV block |
3 |
1 |
— |
— |
|
|
Central Nervous System |
|||||
|
Dizziness |
32 |
19 |
0.4 |
— |
|
|
Headache |
8 |
7 |
0.3 |
— |
|
|
Gastrointestinal |
|||||
|
Diarrhea |
12 |
6 |
0.3 |
— |
|
|
Nausea |
9 |
5 |
— |
— |
|
|
Vomiting |
6 |
4 |
0.1 |
— |
|
|
Metabolic |
|||||
|
Hyperglycemia |
12 |
8 |
0.1 |
— |
|
|
Weight increase |
10 |
7 |
0.1 |
0.5 |
|
|
BUN increased |
6 |
5 |
0.3 |
0.2 |
|
|
NPN increased |
6 |
5 |
0.3 |
0.2 |
|
|
Hypercholesterol- emia |
4 |
3 |
— |
— |
|
|
Musculoskeletal |
|||||
|
Arthralgia |
6 |
5 |
0.1 |
0.2 |
|
|
Resistance Mechanism |
|||||
|
Infection |
2 |
1 |
— |
— |
|
|
Respiratory |
|||||
|
Sinusitis |
5 |
4 |
— |
— |
|
|
Bronchitis |
5 |
4 |
— |
0.2 |
|
|
Urinary/Renal |
|||||
|
Hematuria |
3 |
2 |
— |
— |
|
|
Vision |
|||||
|
Vision abnormal |
5 |
2 |
0.1 |
— |
|
In addition to the events in Table 2, asthenia, chest pain, injury, cardiac failure, syncope, hypertension, abdominal pain, flatulence, anorexia, dyspepsia, palpitation, extrasystoles, gout, hyperkalemia, dehydration, back pain, myalgia, arthritis, angina pectoris, insomnia, depression, anemia, upper respiratory tract infection, viral infection, dyspnea, coughing, , rales, pharyngitis, rhinitis, rash, urinary tract infection, and leg cramps were also reported, but rates were equal to, or greater, in placebo-treated patients.
The following adverse events were reported with a frequency of 1% but £ 2% and more frequently with Coreg in U.S. placebo-controlled trials in patients with mild-to-moderate heart failure:
Incidence >1% to £ 2%
Body as a Whole: Allergy, malaise, hypovolemia.
Cardiovascular: Fluid overload, postural hypotension, aggravated angina pectoris aggravated.
Central and Peripheral Nervous System: Hypesthesia, vertigo, paresthesia
Gastrointestinal: Melena, periodontitis.
Liver and Biliary System: SGPT increased, SGOT increased.
Metabolic and Nutritional: Hyperuricemia, hypoglycemia, hyponatremia, increased alkaline phosphatase, glycosuria, hypervolemia
Platelet, Bleeding and Clotting: Prothrombin decreased, purpura, thrombocytopenia
Psychiatric: Somnolence.
Reproductive, male: Impotence.
Urinary System: renal insufficiency, albuminuria.
Table 3 shows adverse events reported in patients with severe heart failure enrolled in the COPERNICUS trial. Shown are adverse events that occurred more frequently in drug-treated patients than placebo-treated patients with an incidence >2%, regardless of causality. Median study medication exposure was 10.4 months for both carvedilol and placebo patients.
Table 3. Adverse Events (% Occurrence and % Withdrawals) Occurring More Frequently with Coreg than with Placebo in the COPERNICUS trial(Incidence >2%, Regardless of Causality)
|
Adverse Reactions |
Withdrawals |
||||
|
Coreg |
Placebo |
Coreg |
Placebo |
||
|
(n=1156) |
(n=1133) |
(n=1156) |
(n=1133) |
||
|
% occurrence |
% occurrence |
% withdrawals |
% withdrawals |
||
|
Body as a Whole |
|||||
|
Asthenia |
11 |
9 |
0.4 |
0.7 |
|
|
Infection |
3 |
2 |
— |
— |
|
|
Back pain |
3 |
1 |
— |
— |
|
|
Cardiovascular |
|||||
|
Hypotension |
14 |
8 |
0.6 |
0.4 |
|
|
Bradycardia |
10 |
3 |
0.6 |
— |
|
|
Syncope |
8 |
5 |
0.4 |
0.4 |
|
|
Angina pectoris |
6 |
4 |
0.1 |
0.1 |
|
|
Hypertension |
3 |
2 |
— |
0.1 |
|
|
Gastrointestinal |
|||||
|
Diarrhea |
5 |
3 |
0.3 |
— |
|
|
Nausea |
4 |
3 |
— |
0.1 |
|
|
Metabolic and nutritional |
|||||
|
Weight gain |
12 |
11 |
0.1 |
0.1 |
|
|
Peripheral edema |
7 |
6 |
0.2 |
0.1 |
|
|
Generalized edema |
6 |
5 |
0.2 |
0.2 |
|
|
Hyperglycemia |
5 |
3 |
0.0 |
0.1 |
|
|
Hyperkalemia |
3 |
2 |
0.2 |
0.1 |
|
|
Creatinine increased |
3 |
1 |
— |
0.1 |
|
|
Nervous System |
|||||
|
Dizziness |
24 |
17 |
1.3 |
0.6 |
|
|
Headache |
5 |
3 |
— |
0.1 |
|
|
Respiratory |
|||||
|
Upper respiratory infection |
14 |
13 |
0.1 |
— |
|
|
Cough increased |
5 |
4 |
0.1 |
0.2 |
|
|
Rales |
4 |
2 |
0.1 |
— |
|
|
Special senses |
|||||
|
Blurred vision |
3 |
2 |
0.2 |
0.1 |
|
In addition to the events in Table 3, atrial fibrillation, heart failure, peripheral vascular disorder, unstable angina pectoris and ventricular tachycardia,abdominal pain, pain in the extremity, anemia, gout, hypokalemia, dyspnea, bronchitis, lung edema, pneumonia, abnormal kidney function and urinary tract infection were also reported but rates were equal to or greater in placebo patients.
The following adverse events were reported with a frequency of >1% but <2% and more frequently with Coreg the COPERNICUS trial:
Incidence >1% to £ 2%
Cardiovascular: Palpitation, postural hypotension..
Metabolic and Nutritional: Diabetes mellitus, GGT increased, weight loss.
Musculoskeletal: Muscle cramps.
Nervous System: Paresthesia.
Respiratory: Sinusitis.
Urogenital: Kidney failure.
Rates of adverse events were generally similar across demographic subsets (men and women, elderly and non-elderly, blacks and non-blacks).
Hypertension
2nd paragraph and the table revised
Table 2 4 shows adverse events in U.S. placebo-controlled clinical trials for hypertension that occurred with an incidence of greater than 1% regardless of causality, and that were more frequent in drug-treated patients than placebo-treated patients.
|
Adverse Reactions |
Withdrawals |
|||
|
|
Placebo |
|
Placebo |
|
|
Body as a Whole |
||||
|
Fatigue |
4.3 |
3.9 |
0.3 |
0.2 |
|
Injury |
2.9 |
2.6 |
0.1 |
-- |
|
Cardiovascular |
||||
|
Bradycardia |
2.1 |
0.2 |
0.4 |
-- |
|
Postural hypotension |
1.8 |
-- |
1.0 |
-- |
|
Dependent edema |
1.7 |
1.5 |
0.1 |
0.4 |
|
Peripheral edema |
1.4 |
0.4 |
0.2 |
-- |
|
Central Nervous System |
||||
|
Dizziness |
6.2 |
5.4 |
0.4 |
1.3 |
|
Insomnia |
1.6 |
0.6 |
-- |
0.2 |
|
Somnolence |
1.8 |
1.5 |
-- |
-- |
|
Gastrointestinal |
||||
|
Abdominal pain |
1.4 |
1.3 |
0.1 |
-- |
|
Diarrhea |
2.2 |
1.3 |
0.1 |
-- |
|
Hematologic |
||||
|
Thrombocytopenia |
1.1 |
0.2 |
-- |
-- |
|
Metabolic |
||||
|
Hypertriglyceridemia |
1.2 |
0.2 |
-- |
-- |
|
Musculoskeletal |
||||
|
Back pain |
2.3 |
1.5 |
0.1 |
-- |
|
Resistance Mechanism |
||||
|
Viral infection |
1.8 |
1.3 |
-- |
-- |
|
Respiratory |
||||
|
Rhinitis |
2.1 |
1.9 |
-- |
-- |
|
Pharyngitis |
1.5 |
0.6 |
-- |
-- |
|
Dyspnea |
1.4 |
0.9 |
0.4 |
0.2 |
|
Urinary/Renal |
||||
|
Urinary tract infection |
1.8 |
0.6 |
-- |
-- |
Table 4. Adverse Events in U.S. Placebo-Controlled Hypertension Trials
Incidence ³ 1%, Regardless of Causality; Withdrawal Rates due to Adverse Events
|
Adverse Reactions |
Withdrawals |
||||
|
Coreg |
Placebo |
Coreg |
Placebo |
||
|
(n=1,142) |
(n=462) |
(n=1,142) |
(n=462) |
||
|
% occurrence |
% occurrence |
% withdrawals |
% withdrawals |
||
|
Cardiovascular |
|||||
|
Bradycardia |
2 |
— |
0.4 |
— |
|
|
Postural hypotension |
2 |
— |
1.0 |
— |
|
|
Peripheral edema |
1 |
— |
0.2 |
— |
|
|
Central Nervous System |
|||||
|
Dizziness |
6 |
5 |
0.4 |
1.3 |
|
|
Insomnia |
2 |
1 |
— |
0.2 |
|
|
Gastrointestinal |
|||||
|
Diarrhea |
2 |
1 |
0.1 |
— |
|
|
Hematologic |
|||||
|
Thrombocytopenia |
1 |
— |
— |
— |
|
|
Metabolic |
|||||
|
Hypertriglyceri- demia |
1 |
— |
— |
— |
|
|
Resistance Mechanism |
|||||
|
Viral infection |
2 |
1 |
— |
— |
|
|
Respiratory |
|||||
|
Pharyngitis |
2 |
1 |
— |
— |
|
|
Urinary/Renal |
|||||
|
Urinary tract infection |
2 |
1 |
— |
— |
|
In addition to the events in Table 24, abdominal pain, back pain, chest pain, dependent edema, dyspepsia, dyspnea, fatigue, headache, injury, nausea, pain, rhinitis, sinusitis somnolence, and upper respiratory tract infection were also reported, but rates were at least as great in placebo-treated patients.
The following adverse events not described above were reported as possibly or probably related to Coreg in worldwide open or controlled trials with Coreg (carvedilol) in patients with hypertension or congestive heart failure.
deleted
General: Substernal chest pain, edema.
Urinary System: Micturition frequency increased.
Metabolic and Nutritional: Hypokalemia, diabetes mellitus, hypertriglyceridemia
Rates of adverse events were generally similar across demographic subsets (men and women, elderly and non-elderly, blacks and non-blacks).
DOSAGE AND ADMINISTRATION
Congestive Heart Failure
revised
DOSAGE MUST BE INDIVIDUALIZED AND CLOSELY MONITORED BY A PHYSICIAN DURING UP-TITRATION. Prior to initiation of Coreg, the dosing of digitalis, diuretics and ACE inhibitors (if used) should be stabilized it is recommended that fluid retention be minimized. The recommended starting dose of Coreg is 3.125 mg twice daily for two weeks. If this dose is tolerated, it can then be increased to 6.25 mg twice daily. Dosing should then be doubled every 2 weeks to the highest level tolerated by the patient. At initiation of each new dose, patients should be observed for signs of dizziness or light-headedness for one hour. The maximum recommended dose is 25 mg twice daily in patients weighing less than 85 kg (187 lbs) and 50 mg twice daily in patients weighing more than 85 kg. Coreg (carvedilol) should be taken with food to slow the rate of absorption and reduce the incidence of orthostatic effects.
Before each dose increase the patient should be seen in the office and evaluated for symptoms of worsening heart failure, vasodilation (dizziness, light-headedness, symptomatic hypotension) or bradycardia, in order to determine tolerability of Coreg. Transient worsening of heart failure may be treated with increased doses of diuretics although occasionally it is necessary to lower the dose of Coreg or temporarily discontinue it. Symptoms of vasodilation often respond to a reduction in the dose of diuretics or ACE inhibitor. If these changes do not relieve symptoms, the dose of Coreg may be decreased. The dose of Coreg should not be increased until symptoms of worsening heart failure or vasodilation have been stabilized. Initial difficulty with titration should not preclude later attempts to introduce Coreg . If congestive heart failure patients experience bradycardia (pulse rate below 55 beats/min.), the dose of Coreg should be reduced.
Patients who tolerate a dose of 3.125 mg twice daily may have their dose increased to 6.25, 12.5 and 25 mg twice daily over successive intervals of at least two-weeks. Patients should be maintained on lower doses if higher doses are not tolerated. A maximum dose of 50 mg bid has been administered to patients with mild-to-moderate heart failure weighing over 85 kg (187 lbs).
Patients should be advised that initiation of treatment and (to a lesser extent) dosage increases may be associated with transient symptoms of dizziness or lightheadedness (and rarely syncope) within the first hour after dosing. Thus during these periods they should avoid situations such as driving or hazardous tasks, where symptoms could result in injury. In addition, Coreg (carvedilol) should be taken with food to slow the rate of absorption. Vasodilatory symptoms often do not require treatment, but it may be useful to separate the time of dosing of Coreg from that of the ACE inhibitor or to reduce temporarily the dose of the ACE inhibitor. The dose of Coreg should not be increased until symptoms of worsening heart failure or vasodilation have been stabilized
Fluid retention (with or without transient worsening heart failure symptoms) should be treated by an increase in the dose of diuretics.
The dose of Coreg should be reduced if patients experience bradycardia (heart rate <55 beats/min).
Episodes of dizziness or fluid retention during initiation of Coreg can generally be managed without discontinuation of treatment and do not preclude subsequent successful titration of or a favorable response to carvedilol.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
INAPSINE (droperidol) Injection
[November 26, 2001: Akorn]
The following BOX WARNING: is added:
|
WARNING Cases of QT prolongation and/or torsade de pointes have been reported in patients receiving Inapsine at doses at or below recommended doses. Some cases have occurred in patients with no known risk factors for QT prolongation and some cases have been fatal. Due to its potential for serious proarrhythmic effects and death, INAPSINE should be reserved for use in the treatment of patients who fail to show an acceptable response to other adequate treatments, either because of insufficient effectiveness or the inability to achieve an effective dose due to intolerable adverse effects from those drugs (see WARNINGS, ADVERSE REACTIONS, CONTRAINDICATIONS, AND PRECAUTIONS). Cases of QT prolongation and serious arrhythmias (e.g., torsade de pointes) have been reported in patients treated with INAPSINE. Based on these reports, all patients should undergo a 12-lead ECG prior to administration of INAPSINE to determine if a prolonged QT interval (i.e., QTc greater than 440 msec for males or 450 msec for females) is present. If there is a prolonged QT interval, INAPSINE should NOT be administered. For patients in whom the potential benefit of INAPSINE treatment is felt to outweigh the risks of potentially serious arrhythmias, ECG monitoring should be performed prior to treatment and continued for 2-3 hours after completing treatment to monitor for arrhythmias. INAPSINE is contraindicated in patients with known or suspected QT prolongation, including patients with congenital long QT syndrome. INAPSINE should be administered with extreme caution to patients who may be at risk for development of prolonged QT syndrome (e.g., congestive heart failure, bradycardia, use of a diuretic, cardiac hypertrophy, hypokalemia, hypomagnesemia, or administration of other drugs known to increase the QT interval). Other risk factors may include age over 65 years, alcohol abuse, and use of agents such as benzodiazepines, volatile anesthetics, and IV opiates. Droperidol should be initiated at a low dose and adjusted upward, with caution, as needed to achieve the desired effect. |
revised
INAPSINE (droperidol) is indicated:
to produce tranquilization and to reduce the incidence of nausea and vomiting in surgical and diagnostic procedures.
for premedication induction, and as an adjunct in the maintenance of general and regional anesthesia.
in neuroleptanalgesia in which INAPSINE is given concurrently with an opioid analgesic such as SUBLIMAZE® (fentanyl citrate) Injection, to aid in producing tranquility and decreasing anxiety and pain.
INAPSINE (droperidol) is indicated to reduce the incidence of nausea and vomiting associated with surgical and diagnostic procedures.
Revised
INAPSINE (droperidol) is contraindicated in patients with known hypersensitivity to the drug.
INAPSINE is contraindicated in patients with known or suspected QT prolongation (i.e., QTc interval greater than 440 msec for males or 450 msec for females). This would include patients with congenital long QT syndrome.
INAPSINE (droperidol) is contraindicated in patients with known hypersensitivity to the drug.
INAPSINE is not recommended for any use other than for the treatment of perioperative nausea and vomiting in patients for whom other treatments are ineffective or inappropriate (see WARNINGS).
revised
Cases of sudden death have been reported following use of droperidol at high doses (generally 25 mg or greater) in patients at risk for cardiac dysrhythmias due to anoxia, hypercarbia, severe electrolyte disturbances, or alcoholwithdrawal. While these reports do not establish the cause of such death, QT prolongation after INAPSINE administration has been reported and there is at least one case of nonfatal torsades de pointes confirmed by rechallenge.
Because of these reports, INAPSINE is not recommended in the treatment of alcohol withdrawal or in other clinical situations where high doses are likely to be needed in patients at risk for dysrhythmia.
INAPSINE should be administered with extreme caution in the presence of risk factors for development of prolonged QT syndrome, such as: 1) clinically significant bradycardia (less than 50 bpm), 2) any clinically significant cardiac disease, 3) treatment with Class I and Class III antiarrhythmics, 4) treatment with monoamine oxidase inhibitors (MAOI’s), 5) concomitant treatment with other drug products known to prolong the QT interval (see PRECAUTIONS, Drug Interactions), and 6) electrolyte imbalance, in particular hypokalemia and hypomagnesemia, or concomitant treatment with drugs (e.g. diuretics) that may cause electrolyte imbalance.
Effects on Cardiac Conduction
A dose-dependent prolongation of the QT interval was observed within 10 minutes of droperidol administration in a study of 40 patients without known cardiac disease who underwent extracranial head and neck surgery. Significant QT prolongation was observed at all three dose levels evaluated, with 0.1, 0.175, and 0.25 mg/kg associated with prolongation of median QTc by 37, 44, and 59 msec, respectively.
Cases of QT prolongation and serious arrhythmias (e.g. torsade de pointes, ventricular arrythmias, cardiac arrest, and death) have been observed during post-marketing treatment with INAPSINE. Some cases have occurred in patients with no known risk factors and at doses at or below recommended doses. There has been at least one case of nonfatal torsade de pointes confirmed by rechallenge.
Based on these reports, all patients should undergo a 12-lead ECG prior to administration of INAPSINE to determine if a prolonged QT interval (i.e., QTc greater than 440 msec for males or 450 msec for females) is present. If there is a prolonged QT interval, INAPSINE should NOT be administered. For patients in whom the potential benefit of INAPSINE treatment is felt to outweigh the risks of potentially serious arrhythmias, ECG monitoring should be performed prior to treatment and continued for 2-3 hours after completing treatment to monitor for arrhythmias.
FLUIDS AND OTHER COUNTERMEASURES TO MANAGE HYPOTENSION SHOULD BE READILY AVAILABLE.
As with other CNS depressant drugs, patients who have received INAPSINE (droperidol) should have appropriate surveillance.
It is recommended that opioids, when required, initially be used in reduced doses. As with other neuroleptic agents, very rare reports of neuroleptic malignant syndrome (altered consciousness, muscle rigidity and autonomic instability) have occurred in patients who have received INAPSINE (droperidol).
Since it may be difficult to distinguish neuroleptic malignant syndrome from malignant hyperpyrexia in the perioperative period, prompt treatment with dantrolene should be considered if increases in temperature, heart rate or carbon dioxide production occur.
PRECAUTIONS:
General
fourth paragraph revised
Vital signs and ECG should be monitored routinely.
added
Potentially Arrhythmogenic Agents: Any drug known to have the potential to prolong the QT interval should not be used together with INAPSINE. Possible pharmacodynamic interactions can occur between INAPSINE and potentially arrhythmogenic agents such as class I or III antiarrhythmics, antihistamines that prolong the QT interval, antimalarials, calcium channel blockers, neuroleptics that prolong the QT interval, and antidepressants.
Caution should be used when patients are taking concomitant drugs known to induce hypokalemia or hypomagnesemia as they may precipitate QT prolongation and interact with INAPSINE. These would include diuretics, laxatives and supraphysiological use of steroid hormones with mineralocorticoid potential.
CNS Depressant Drugs: Other CNS depressant drugs (e.g., barbiturates, tranquilizers, opioids, and general anesthetics) have additive or potentiating effects with INAPSINE. When patients have received such drugs, the dose of INAPSINE required will be less than usual. Following the administration of INAPSINE, the dose of other CNS depressant drugs should be reduced.
added
QT interval prolongation, torsade de pointes, cardiac arrest, and ventricular tachycardia have been reported in patients treated with INAPSINE. Some of these cases were associated with death. Some cases occurred in patients with no known risk factors, and some were associated with droperidol doses at or below recommended doses.
Physicians should be alert to palpitations, syncope, or other symptoms suggestive of episodes of irregular cardiac rhythm in patients taking INAPSINE and promptly evaluate such cases (see WARNINGS, Effects on Cardiac Conduction).
1st paragraph revised
Manifestations: The manifestations of INAPSINE (droperidol) overdosage are an extension of its pharmacologic actions and may include QT prolongation and serious arrhythmias (e.g., torsade de pointes) (see BOX WARNING, WARNINGS, and PRECAUTIONS).
4th paragraph revised
The intravenous LD50 Median Lethal Dose of INAPSINE is 20-43 mg/kg in mice; 30 mg/kg in rats: 25 mg/kg in dogs and 11-13 mg/kg in rabbits. The intramuscular intravenous LD50 Median Lethal Dose of INAPSINE is 195 mg/kg in mice; 104-110 mg/kg in rats; 97 mg/kg in rabbits and 200 mg/kg in guinea pigs.
Revised
Vital signs and ECG should be monitored routinely.
Usual Adult Dosage
I. Premedication – to be appropriately modified in the elderly, debilitated and those who have received other depressant drugs) 2.5 mg (1 mL) may be administered intramuscularly 30 to 60 minutes preoperatively.
II. Adjunct to General Anesthesia -
Induction – 2.5 mg (1 mL) may be administered (usually intravenously) along with an analgesic and/or general anesthetic. Smaller doses may be adequate. The total amount of INAPSINE administered should be titrated to obtain the desired effect based on the individual patient’s response.
Maintenance – 1.25 to 2.5 mg (0.5 to 1 mL) usually intravenously.
III. Use without a general anesthetic in diagnostic procedures – Administer the usual I.M. premedication 2.5 mg (1 mL) 30 to 60 minutes before procedure. Additional 1.25 mg (0.5 mL) amounts of INAPSINE may be administered, usually intravenously.
NOTE: When INAPSINE is used in certain procedures, such as bronchoscopy, appropriate topical anesthesia is still necessary.
IV. Adjunct to regional anesthesia – 2.5 mg (1 mL) may be administered intramuscularly or slowly intravenously when additional sedation is required.
Usual Children's Dosage
For children two to twelve years of age, a 1.0 mg (0.4 ml) is recommended for premedication or for induction of anesthesia.
Adult dosage: The maximum recommended initial dose of Inapsine is 2.5 mg IM or slow IV. Additional 1.25 mg doses of INAPSINE may be administered to achieve the desired effect. However, additional doses should be administered with caution, and only if the potential benefit outweighs the potential risk.
Children’s dosage: For children two to 12 years of age, the maximum recommended initial dose is 0.1 mg/kg, taking into account the patient’s age and other clinical factors. However, additional doses should be administered with caution and only if the potential benefit outweighs the potential risk.
See WARNINGS and PRECAUTIONS for use of INAPSINE with other CNS depressants and in patients with altered response.
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. If such abnormalities are observed, the drug should not be administered.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
INDERIDE (propranolol and hydrochlorothiazide)
Tablets
INDERIDE LA (propranolol and hydrochlorothiazide) Long-Acting Capsules
[November 1, 2001: Wyeth-Ayerst]
[Other labeling changes not found in 2001 PDR: http://www.fda.gov/medwatch/SAFETY/2001/may01.htm#inderi]
WARNINGS
Propranolol Hydrochloride
Added
Acute increases in blood pressure have occurred after insulin-induced hypoglycemia in patients on propranolol.
PRECAUTIONS
added
Geriatric Use
Clinical studies of Inderide (or Inderide LA) did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
Nursing Mothers
Hydrochlorothiazide
added
Thiazides appear in breast milk. If use of the drug is deemed essential, the patient should stop nursing.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
Lithium Carbonate Capsules and Lithium Citrate Syrup
[November 26, 2001: Roxane]
Contact sponsor for the details.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
LUPRON (leuprolide acetate) DEPOT-and DEPOT
3
[November 14, 2001: Tap Pharmaceutical]
[other safety-related labeling changes not found in 2001 PDR:
http://www.fda.gov/medwatch/SAFETY/2001/sep01.htm#lupron]
PRECAUTIONS
Information for Patients
Added as #5
Patients should be counseled on the possibility of the development or worsening of depression and the occurrence of memory disorders.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
MYCOBUTIN (rifabutin) Capsules
[November 9, 2001: Pharmacia & Upjohn]
CLINICAL PHARMACOLOGY
Pharmacokinetics
new subheadings added and the text re-ordered and revised
Absorption:Following a single oral dose of 300 mg to nine healthy adult volunteers, rifabutin was readily absorbed from the gastrointestinal tract with mean (+/-SD) peak plasma levels (Cmax) of 375 (+/-267) ng/mL (range: 141 to 1033 ng/mL) attained in 3.3 (+/-0.9) hours (Tmax range: 2 to 4 hours). Absolute bioavailability assessed in five HIV-positive patients, who received both oral and intravenous doses, averaged 20%. Total recovery of radioactivity in the urine indicates that at least 53% of the orally administered rifabutin dose is absorbed from the gastrointestinal tract. The bioavailability of rifabutin from the capsule dosage form, relative to an oral solution, was 85% in 12 healthy adult volunteers. High-fat meals slow the rate without influencing the extent of absorption from the capsule dosage form. Plasma concentrations post-Cmax declined in an apparent biphasic manner. kinetic Pharmacokinetic dose-proportionality was has been established over the 300 to 600 mg dose range in nine healthy adult volunteers (crossover design) and in 16 early symptomatic human immunodeficiency virus (HIV)-positive patients over a 300 to 900 mg dose range.
Distribution: Rifabutin, Due to its high lipophilicity, rifabutin demonstrates a high propensity for distribution and intracellular tissue uptake. Estimates of apparent steady-state distribution volume (9.3 ± 1.5 L/kg) in five HIV-positive patients, following I.V. dosing, exceed total body water by approximately 15-fold. Substantially higher intracellular tissue levels than those seen in plasma have been observed in both rat and man. . Following intravenous dosing, estimates of apparent steady-state distribution volume (9.3 +/- 1.5 L/kg) in five HIV-positive patients exceeded total body water by approximately 15-fold. Substantially higher intracellular tissue levels than those seen in plasma have been observed in both rat and man. The lung to plasma concentration ratio, obtained at 12 hours, was found to be approximately 6.5 in four surgical patients administered an oral dose. Mean rifabutin steady-state trough levels (Cp,min SS ; 24-hour post-dose) ranged from 50 to 65 ng/mL in HIV-positive patients and in healthy adult volunteers. About 85% of the drug is bound in a concentration-independent manner to plasma proteins over a concentration range of 0.05 to 1 mg/mL. Binding does not appear to be influenced by renal or hepatic dysfunction. Rifabutin was slowly eliminated from plasma in seven healthy adult volunteers, presumably because of distribution-limited elimination, with a mean terminal half-life of 45 (+/- 17) hours (range: 16 to 69 hours). Although the systemic levels of rifabutin following multiple dosing decreased by 38%, its terminal half-life remained unchanged.
Metabolism: Of the five metabolites that have been identified, 25-O-desacetyl and 31-hydroxy are the most predominant, and show a plasma metabolite:parent area under the curve ratio of 0.10 and 0.07, respectively. The former has an activity equal to the parent drug and contributes up to 10% to the total antimicrobial activity.
Excretion:"Mean systemic clearance (CLS/F) in healthy adult volunteers following a single oral dose was 0.69 (±0.32) L/hr/kg (range: 0.46 to 1.34 L/hr/kg). Renal and biliary clearance of unchanged drug each contribute approximately 5% to CLS/F. About 30% of the dose is excreted in the feces. A mass-balance study in three healthy adult volunteers with 14 C-labeled drug has shown that 53% of the oral dose was excreted in the urine, primarily as metabolites.
"A mass-balance study in three healthy adult volunteers with 14C-labeled rifabutin showed that 53% of the oral dose was excreted in the urine, primarily as metabolites. About 30% of the dose is excreted in the feces. Mean systemic clearance (CLs/F) in healthy adult volunteers following a single oral dose was 0.69 (+/- 0.32) L/hr/kg (range: 0.46 to 1.34 L/hr/kg). Renal and biliary clearance of unchanged drug each contribute approximately 5% to CLs/F."
"No rifabutin disposition information is currently available in children or adolescents under 18 years of age."
·Pharmacokinetics in Special Populations:
Geriatric:
Compared to healthy volunteers, steady-state kinetics of MYCOBUTIN are more variable in elderly patients (>70 years).
Pediatric: The pharmacokinetics of MYCOBUTIN have not been studied in subjects under 18 years of age.
Renal Insufficiency: The disposition of rifabutin (300 mg) was studied in 18 patients with varying degrees of renal function. Area under plasma concentration time curve (AUC) increased by about 71% in patients with severe renal insufficiency (creatinine clearance below 30 mL/min) compared to patients with creatinine clearance (Crcl) between 61-74 mL/min. In patients with mild to moderate renal insufficiency (Crcl between 30-61 mL/min), the AUC increased by about 41%. A reduction in the dosage of rifabutin is recommended for patients with Crcl < 30 mL/min (see DOSAGE AND ADMINISTRATION).
Drug-Drug Interactions (see also PRECAUTIONS-Drug Interactions)
Rifabutin induces the enzymes of the cytochrome P450 3A subfamily (CYP3A) and therefore may reduce the plasma concentrations of drugs that are principally metabolized by those enzymes. Rifabutin is also metabolized by CYP3A. Thus, some drugs that inhibit CYP3A may significantly increase plasma concentrations of rifabutin.
Antifungals:
Fluconazole: Fluconazole (200 mg/day for 2 weeks) increased the AUC of rifabutin (300 mg/day for 2 weeks) by 82% and Cmax by 88% in 12 HIV-infected patients who were on zidovudine (500 mg/day) maintenance therapy (see PRECAUTIONS-Drug Interactions). Rifabutin did not affect the pharmacokinetics of fluconazole.
Itraconazole: Coadministration of itraconazole (200 mg/day) with rifabutin (300 mg/day) in six HIV-infected patients reduced both the AUC and Cmax of itraconazole by 70% to 75% (see PRECAUTIONS-Drug Interactions).
Antipneumocystis Agents:
Dapsone: Rifabutin (300 mg/day) decreased the AUC of dapsone (50 mg/day) in HIV-infected patients (n=16) by about 27% to 40%.
Sulfamethoxazole-trimethoprim: Coadministration of rifabutin (300 mg/day) and sulfamethoxazole-trimethoprim (double strength) in 12 HIV-infected patients decreased the AUC of sulfamethoxazole-trimethoprim by about 15% to 20%. When trimethoprim was given alone, the AUC of trimethoprim was decreased by 14% and the Cmax by 6%. Sulfamethoxazole-trimethoprim did not alter the pharmacokinetics of rifabutin.
Antiretroviral Agents:
Delavirdine: In 7 HIV-infected patients, rifabutin (300 mg/day) decreased delavirdine (400 mg q 8h) AUC by about 80%, Cmax by about 75%, and mean trough plasma concentrations by about 95%. Based on comparisons with historical data, delavirdine appeared to increase the AUC of rifabutin by at least 100% (see PRECAUTIONS-Drug Interactions).
Didanosine: In 12 HIV-infected patients, coadministration of rifabutin (300 or 600 mg/day) and didanosine (167-375 mg BID) did not alter the pharmacokinetics of either drug.
Indinavir: In healthy volunteers, coadministration of indinavir (800 mg q 8h) and rifabutin (300 mg/day) decreased the AUC of indinavir by about 30% and increased the AUC of rifabutin by about 200% (see PRECAUTIONS-Drug Interactions).
Nelfinavir: Coadministration of nelfinavir (750 mg q 8h for 8 days) and rifabutin (300 mg/day for 7-8 days) decreased the AUC and Cmax of nelfinavir by about 32% and 25%, respectively, and increased the AUC and Cmax of rifabutin by about 207% and 146%, respectively (see PRECAUTIONS-Drug Interactions).
Ritonavir: Coadministration of ritonavir (500 mg q 12h) and rifabutin (150 mg/day) increased the AUC and Cmax of rifabutin by more than 400% and 250%, respectively (see PRECAUTIONS-Drug Interactions).
Saquinavir: In 12 HIV-infected patients, rifabutin (300 mg/day) decreased the AUC of saquinavir (600 mg TID) by about 40% (see PRECAUTIONS-Drug Interactions).
Zidovudine: In 16 HIV-infected patients, on zidovudine (100 or 200 mg q 4h), rifabutin (300 or 450 mg/day) lowered the Cmax and AUC of zidovudine by about 48% and 32%, respectively. However, zidovudine levels remained within the therapeutic range during coadministration of rifabutin. Zidovudine did not affect the pharmacokinetics of rifabutin.
Antituberculosis Agents:
In studies conducted in healthy volunteers, rifabutin (300 mg) did not alter the pharmacokinetics of ethambutol (n=10) or isoniazid (n=10).
Macrolides:
Clarithromycin: In studies conducted in HIV-infected patients, coadministration of rifabutin (300 mg/day) and clarithromycin (500 mg q 12h) decreased the AUC of clarithromycin by about 50% (n=12) and increased the AUC of rifabutin by about 75% (n=14) (see PRECAUTIONS-Drug Interactions).
Other Drugs:
Methadone: Rifabutin did not alter the pharmacokinetics of methadone in 24 HIV-infected, methadone-maintained, former intravenous drug users.
Oral contraceptives: In 22 healthy female volunteers receiving an oral contraceptive (35 mcg ethinylestradiol (EE) and 1 mg norethindrone (NE) daily for 21 days, rifabutin decreased EE (AUC) and Cmax by 35% and 20%, respectively, and NE AUC by 46% (see PRECAUTIONS-Drug Interactions).
Theophylline: Rifabutin did not alter the pharmacokinetics of theophylline when coadministered in 11 healthy volunteers.
Other drugs: The structurally similar drug, rifampin, is known to reduce the plasma concentrations of a number of other drugs (see prescribing information for rifampin). Although rifabutin is a weaker enzyme inducer than rifampin, rifabutin may be expected to have some effect on those drugs as well.
PRECAUTIONS
Drug Interactions
revised
In 10 healthy adult volunteers and 8 HIV-positive patients, steady-state plasma levels of zidovudine (ZDV), an antiretroviral agent which is metabolized mainly through glucuronidation, were decreased after repeated dosing with MYCOBUTIN; the mean decrease in Cmax and AUC was decreased by 48% and 32%, respectively. In vitro studies have demonstrated that rifabutin does not affect the inhibition of HIV by ZDV. Steady-state kinetics in 12 HIV-positive patients show that both the rate and extent of systemic availability of didanosine (ddl), was not altered after repeated dosing of MYCOBUTIN. Rifabutin has liver enzyme-inducing properties. The related drug rifampin is known to reduce the activity of a number of other drugs, including dapsone, narcotics (including methadone), anticoagulants, corticosteroids, cyclosporine, cardiac glycoside preparations, quinidine, oral contraceptives, oral hypoglycemic agents (sulfonylureas), and analgesics. Rifampin has also been reported to decrease the effects of concurrently administered ketoconazole, barbiturates diazepam, verapamil, beta-adrenergic blockers, clofibrate, progestins, disopyramide, mexiletine theophylline, chloramphenicol, and anticonvulsants. Because of the structural similarity of rifabutin and rifampin, MYCOBUTIN may be expected to have some effect on these drugs as well. However, unlike rifampin, MYCOBUTIN appears not to affect the acetylation of isoniazid. When rifabutin was compared with rifampin in a study with 8 healthy normal volunteers, rifabutin appeared to be a less potent enzyme inducer than rifampin. The significance of this finding for clinical drug interactions is not known. Dosage adjustment of drugs listed above may be necessary if they are given concurrently with MYCOBUTIN. Patients using oral contraceptives should consider changing to nonhormonal methods of birth control.
Effects on Other Drugs: Rifabutin induces CYP3A enzymes and therefore may reduce the plasma concentrations of drugs metabolized by those enzymes. This effect may reduce the efficacy of standard doses of such drugs, which include itraconazole, clarithromycin, and saquinavir (see CLINICAL PHARMACOLOGY-Drug-Drug Interactions).
Effects on Rifabutin: Some drugs that inhibit CYP3A may significantly increase the plasma concentration of rifabutin. Because high plasma levels of rifabutin may increase the risk of adverse reactions, carefully monitor patients receiving coadministration of such drugs, which include fluconazole and clarithromycin (see CLINICAL PHARMACOLOGY-Drug-Drug Interactions). In some cases, the dosage of MYCOBUTIN may need to be reduced when it is coadministered with such a drug (see below).
Antiretrovirals:
Delavirdine: Coadministration of rifabutin and delavirdine is not recommended because rifabutin substantially decreases the plasma concentrations of delavirdine, and delavirdine increases the plasma concentrations of rifabutin (see CLINICAL PHARMACOLOGY-Drug-Drug Interactions).
Indinavir: Coadministration of indinavir and rifabutin increases the plasma concentration of rifabutin. In patients receiving coadministration of indinavir, reduce the dosage of MYCOBUTIN by half (see CLINICAL PHARMACOLOGY-Drug-Drug Interactions).
Nelfinavir: Coadministration of nelfinavir increases the plasma concentration of rifabutin. In patients receiving nelfinavir, reduce the dosage of MYCOBUTIN by half (see CLINICAL PHARMACOLOGY-Drug-Drug Interactions).
Ritonavir: Coadministration of ritonavir is not recommended because it substantially increases the plasma concentration of rifabutin (see CLINICAL PHARMACOLOGY-Drug-Drug Interactions). High plasma concentrations of rifabutin may increase the risk of adverse reactions, including uveitis.
Other Drugs:
Oral contraceptives: Rifabutin may decrease the efficacy of oral contraceptives by inducing drug metabolism of ethinylestradiol and norethindrone. Women using oral contraceptives should be advised to change to or supplement with nonhormonal methods of birth control during treatment with MYCOBUTIN.
Other drugs: The structurally similar drug, rifampin, is known to reduce the plasma concentrations of a number of other drugs (see prescribing information for rifampin). Although rifabutin is a weaker enzyme inducer than rifampin, it may be expected to have some effect on those drugs as well.
Pediatric Use
revised statement is added at the end.
In addition, corneal deposits have been observed in some patients during routine ophthalmologic surveillance of HIV-positive pediatric patients receiving MYCOBUTIN as part of a multiple-drug regimen for MAC prophylaxis. These are tiny, almost transparent, asymptomatic peripheral and central corneal deposits which do not impair vision.
added
Geriatric Use
Clinical studies of MYCOBUTIN did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy (see CLINICAL PHARMACOLOGY).
DOSAGE AND ADMINISTRATION
added
For patients with severe renal impairment (creatinine clearance less than 30mL/min), the dose of MYCOBUTIN should be reduced by 50%. No dosage adjustment is required for patients with mild to moderate renal impairment. Reduction of the dose of MYCOBUTIN may also be needed for patients receiving concomitant treatment with certain other drugs (see PRECAUTIONS-Drug Interactions).
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
PRIMAXIN (imipenem and cilastatin) I.M. and I.V.
November 30, 2001: Merck
Added
PRIMAXIN IV
CLINICAL PHARMACOLOGY
In healthy elderly volunteers (65 to 75 years of age with normal renal function for their age), the pharmacokinetics of a single dose of imipenem 500 mg and cilastatin 500 mg administered intravenously over 20 minutes are consistent with those expected in subjects with slight renal impairment for which no dosage alteration is considered necessary. The mean plasma half-lives of imipenem and cilastatin are 91± 7.0 minutes and 69 ± 15 minutes, respectively. Multiple dosing has no effect on the pharmacokinetics of either imipenem or cilastatin, and no accumulation of imipenem/cilastatin is observed.
PRECAUTIONS
Geriatric Use
Of the approximately 3600 subjects >18 years of age in clinical studies, including post-marketing studies, of PRIMAXIN I.V., approximately 2800 received PRIMAXIN I.V. Of the subjects who received PRIMAXIN I.V., data are available on approximately 800 subjects who were 65 and over, including approximately 300 subjects who were 75 and over. No overall differences in safety or effectiveness were observed between these subjects and younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
This drug is known to be substantially excreted by the kidney, and the risk of toxic reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection, and it may be useful to monitor renal function.
No dosage adjustment is required based on age (see CLINICAL PHARMACOLOGY, Adults). Dosage adjustment in the case of renal impairment is necessary (see DOSAGE AND ADMINISTRATION, Reduced Intravenous Schedule for Adults with Impaired Renal
Function and/or Body Weight < 70 kg).
PRIMAXIN IM
PRECAUTIONS
Geriatric Use
Clinical studies of PRIMAXIN I.M. did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects; however, clinical studies of PRIMAXIN I.V. in a sufficient number of subjects aged 65 and over have not revealed overall differences in safety or effectiveness between these subjects and younger subjects (refer to the package circular for PRIMAXIN I.V.). Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
This drug is known to be substantially excreted by the kidney, and the risk of toxic reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection, and it may be useful to monitor renal function. Dosage adjustment in the case of renal impairment is necessary (see DOSAGE AND ADMINISTRATION, ADULTS WITH IMPAIRED RENAL FUNCTION).
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
PROSOL 20% (amino acids) Sulfite Free Injectable
[November 14, 2001: Baxter Healthcare]
Pediatric Use
revised
Safety and effectiveness of 20% ProSol™ - sulfite-free (Amino Acid)
Injection solution
have not been established by adequate and well controlled
studies in pediatric patients have
not been established.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
REGLAN (metoclopromide) Tablets, Injection and Syrup
[November 7, 2001: AH Robins]
DESCRIPTION:
Added for Reglan Injection
This product is light sensitive. It should be inspected before use and discarded if either color or particulate is observed."
PRECAUTIONS
General
added after the second paragraph
Because metoclopramide produces a transient increase in plasma aldosterone, certain patients, especially those with cirrhosis or congestive heart failure, may be at risk of developing fluid retention and volume overload. If these side effects occur at any time during metoclopramide therapy, the drug should be discontinued."
second sentence of the last paragraph revised
Although adverse events related to this possibility have not been reported to date, the This possibility should be considered and weighed when deciding whether to use metoclopramide or nasogastric suction in the prevention of postoperative nausea and vomiting."
ADVERSE REACTIONS
Cardiovascular:
Hypotension…fluid retention, acute congestive heart failure, and possible AV block (see CONTRAINDICATIONS and PRECAUTIONS).
HOW SUPPLIED
added after the paragraph which begins,
"Store vials and ampuls in carton until used…" "This product is light sensitive. It should be inspected before use and discarded if either color or particulate is observed."
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
ROCEPHIN (ceftriaxone injection) in D5W Galaxy Container
[November 21, 2001: Hoffman LaRoche]
Revisions same as that approved August 25, 2000 for Rocephin (ceftriaxone injection)
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
SINGULAIR (montelukast Na) Chewable Tablets
[November 23, 2001: Merck]
[Other labeling changes not found in 2001 PDR: http://www.fda.gov/medwatch/SAFETY/2001/aug01.htm#singul]
CLINICAL PHARMACOLOGY
Clinical Studies
PEDIATRIC PATIENTS 2 TO 5 YEARS OF AGE
The efficacy of SINGULAIR for the chronic treatment of asthma in pediatric patients 2 to 5 years of age was explored in a 12- week placebo- controlled safety and tolerability study in 689 patients, 461 of whom were treated with SINGULAIR. While the primary objective was to determine the safety and tolerability of SINGULAIR in this age group, the study included exploratory efficacy evaluations, including daytime and overnight asthma symptom scores, b - agonist use, oral corticosteroid rescue, and the physician’s global evaluation. The findings of these exploratory efficacy evaluations, along with pharmacokinetics and extrapolation of efficacy data from older patients, support the overall conclusion that SINGULAIR is efficacious in the maintenance treatment of asthma in patients 2 to 5 years of age.
PRECAUTIONS
Pediatric Use
The safety of SINGULAIR 4-mg chewable tablets in pediatric patients 2 to 5
years of age has been demonstrated in an interim
analysis of 314 pediatric patients in a 12-week double-blind, placebo-controlled
study in approximately 650 patients by adequate and well-controlled
data (see ADVERSE REACTIONS ). The efficacy
of SINGULAIR is supported in this age group
is extrapolated by
extrapolation from the demonstrated efficacy in adolescent
and adult patients 15
6 years of age and older and
pediatric patients 6 to 14 years of age with asthma based and
is based on similar mean systemic exposure
(AUC) pharmacokinetic data,
and as well as the assumption that the disease
course, pathophysiology and the drug's effect are substantially similar among
these populations. Efficacy in this age group is further
supported by efficacy assessments from a large, well- controlled safety study
conducted in patients 2 to 5 years of age.
ADVERSE REACTIONS
Pediatric Patients 2 to 5 Years of Age
revised
Safety data for SINGULAIR in pediatric patients 2 to 5 years of age are available
from an interim analysis of 314 pediatric patients from a 12-week, double-blind,
placebo-controlled clinical study in approximately 650 patients. The safety
profile of SINGULAIR in this interim analysis of patients who received SINGULAIR
for at least 6 weeks was generally similar to the safety profile in pediatric
patients 6 to 14 years of age. SINGULAIR has been evaluated
for safety in 573 pediatric patients 2 to 5 years of age in single and multiple
dose studies. Cumulatively, 426 pediatric patients 2 to 5 years of age were
treated with SINGULAIR for at least 3 months, 230 for 6 months or longer, and
63 patients for one year or longer in clinical trials. SINGULAIR
4 mg administered once daily at bedtime was generally well tolerated in clinical
trials.In pediatric patients 2 to 5 years of age receiving SINGULAIR, the following
events occurred with a frequency >2% and more frequently than in pediatric
patients who received placebo, regardless of causality assessment: fever,
cough, abdominal pain, diarrhea, headache, rhinorrhea, sinusitis,
otitis,influenza, bronchitis,
leg pain, thirst, sneezing rash, ear
pain, gastroenteritis, eczema, urticaria, varicella,
pneumonia, dermatitis, and conjunctivitis.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
TAMIFLU (oseltamivir phosphate) Capsules and Oral Suspension
[November 5, 2001: Hoffman LaRoche]
[Other labeling changes not found in 2001 PDR: http://www.fda.gov/medwatch/SAFETY/2001/jul01.htm#tamifl ]
ADVERSE REACTIONS:
Observed During Clinical Practice for Treatment
revised
General: Rash, swelling of the face or tongue, toxic epidermal necrolysis
Digestive: Hepatitis, liver function tests abnormal
Cardiac: Arrhythmia
Neurologic: Seizure, confusion
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
[November 30, 2001: Merck]
PRECAUTIONS
added
Geriatric Use
No overall differences in safety or effectiveness have been observed between elderly and younger patients.
ADVERSE REACTIONS
Hypersensitivity
revised
Signs and symptoms of systemic allergic reactions, including anaphylaxis, angioedema, urticaria, and localized and generalized rash.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
VASOTEC (enalapril maleate) I.V. and
Tablets
VASERETIC (enalapril maleate/hydrochlorothiazide) Tablets
November 27, 2001: Merck
[Other labeling changes not found in 2001 PDR: http://www.fda.gov/medwatch/SAFETY/2001/feb01.htm#vasote]
CLINICAL PHARMACOLOGY
Pharmacodynamics
added at the end of the last paragraph:
(See PRECAUTIONS, Drug Interactions, Enalapril Maleate)
PRECAUTIONS
Drug Interactions/Enalapril Maleate/Non-steroidal Anti-inflammatory Agents
A second paragraph has been added:
In a clinical pharmacology study, indomethacin or sulindac was administered to hypertensive patients receiving enalapril maleate. In this study there was no evidence of a blunting of the antihypertensive action of enalapril maleate. However, reports suggest that NSAIDs may diminish the antihypertensive effect of ACE-inhibitors. This interaction should be given consideration in patients taking NSAIDs concomitantly with ACE-inhibitors.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
VIDEX (didanosine) Buffered Tablets,
Powder for Oral Solution, Pediatric Powder
[November 16, 2001: Bristol-Myers]
[Other safety-related information: http://www.fda.gov/medwatch/safety/2001/safety01.htm#zerit]
BOXED WARNING, WARNINGS, and PRECAUTIONS sections revised to include the following
Fatal lactic acidosis has been reported in pregnant women who received the combination of didanosine and stavudine with other antiretroviral agents." And "The combination of didanosine and stavudine should be used with caution during pregnancy and is recommended only if the potential benefit clearly outweighs the potential risks.
PRECAUTIONS
Added as third paragrapgh
Fat Redistribution
Redistribution/accumulation of body fat including central obesity, dorsocervical fat enlargement (buffalo hump), peripheral wasting, facial wasting, breast enlargement, and "cushingoid appearance" have been observed in patients receiving antiretroviral therapy. The mechanism and long-term consequences of these events are currently unknown. A causal relationship has not been established.
Information for patients
Added as last paragraph
Patients should be informed that redistribution or accumulation of body fat may occur in patients receiving antiretroviral therapy and that the cause and long-term health effects of these conditions are not known at this time.
ADVERSE REACTIONS
Observed During Clinical Practice
revised
Body as a Whole - alopecia, anaphylactoid reaction, asthenia, chills/fever, pain, and redistribution/accumulation of body fat (see PRECAUTIONS: Fat Redistribution).
Patient information leaflet
Other side effects:
revised
The most common side effects in adults taking VIDEX are diarrhea, neuropathy (nerve disorders), chills or fever, rash, abdominal pain, weakness, headache, and nausea and vomiting. Children may have similar side effects as adults.
Changes in body fat have been seen in some patients taking antiretroviral therapy. These changes may include increased amount of fat in the upper back and neck ("buffalo hump"), breast, and around the trunk. Loss of fat from the legs, arms, and face may also happen. The cause and long-term health effects of these conditions are not known at this time.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
WYTENSIN ( guanabenz acetate) Tablets
[November 27, 2001: Wyeth Ayerst]
PRECAUTIONS
Added
Geriatric Use
Clinical studies of Wytensin did not include sufficient number of subject aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients.
In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
ZOCOR (simvastatin) Tablet
[November 14, 2001: Merck]
INDICATION AND USAGE
[Table 4 revised. New table shown below:]
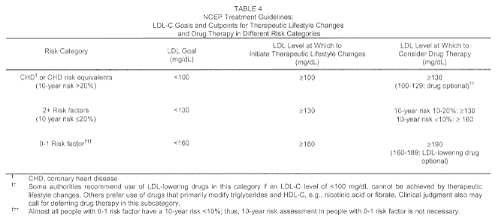
After the LDL-C goal has been achieved, if the TG is still >200 mg/dL, non-HDL-C (total-C minus HDL-C) becomes a secondary target of therapy. Non-HDL-C goals are set 30 mg/dL higher than LDL-C goals for each risk category.
WARNINGS
Skeletal Muscle
added
The risk of myopathy appears to be increased by concomitant administration of verapamil (see PRECAUTIONS, Drug Interactions). In an analysis of clinical trials involving 25,248 patients treated with simvastatin 20 to 80 mg, the incidence of myopathy was higher in patients receiving verapamil and simvastatin (4/635; 0.63%) than in patients taking simvastatin without a calcium channel blocker (13 patients/21,224, 0.061%)."
PRECAUTIONS
Drug Interactions
added:
"The risk of myopathy appears to be increased by concomitant administration of verapamil."
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
ZOVIRAX (acyclovir) Tablets, Capsules, Suspension
[November 14, 2001: GlaxoSmithKline]
[Other labeling changes not found in 2001 PDR: http://www.fda.gov/medwatch/SAFETY/2001/may01.htm#zovira ]
VIROLOGY
Mechanism of Antiviral Action:
The last sentence in the first paragraph deleted: "In cell culture, acyclovir’s highest antiviral activity is against HSV-1, followed in decreasing order of potency against HSV-2 and VZV."
Drug Resistance
first sentence revised. "Resistance of VZV to antiviral nucleoside analogues acyclovir can result from qualitative or quantitative changes in the viral TK and/or DNA polymerase."
CLINICAL PHARMACOLOGY
Special Populations,
Geriatrics section added:
Geriatrics: Acyclovir plasma concentrations are higher in geriatric patients compared to younger adults, in part due to age-related changes in renal function. Dosage reduction may be required in geriatric patients with underlying renal impairment (see PRECAUTIONS: Geriatric Use)."
PRECAUTIONS
Carcinogenesis, Mutagenesis, Impairment of Fertility
first sentence of the first paragraph revised
"The data presented below include references to peak steady-state plasma acyclovir concentrations observed in humans treated with 800 mg given orally 6 5 times a day (dosing appropriate for treatment of herpes zoster) or 200 mg given orally 6 5 times a day (dosing appropriate for treatment of genital herpes)."
third paragraph revised
"Acyclovir was tested in 16 genetic in vitro
and in vivo toxicity assays. No evidence
of mutagenicity was observed in 4 microbial assays. ). Acyclovir
was positive in 5 of the assays."Acyclovir
demonstrated mutagenic activity in 2 in vitro cytogenetic assays (1 mouse lymphoma
cell line and human lymphocytes No mutagenic activity was observed in 5 in vitro
cytogenetic assays (3 Chinese hamster ovary cell lines and 2 mouse lymphoma
cell lines)."
deleted: Paragraphs 4 & 5
"A positive result was demonstrated in 1 of 2 in vitro cell transformation assays, and morphologically transformed cells obtained in this assay formed tumors when inoculated into immunosuppressed, syngeneic, weanling mice. No activity was demonstrated in another, possibly less sensitive, in vitro cell transformation assay."
Paragraph 5
"Acyclovir caused chromosomal damage in Chinese hamsters at 380 to 760 times
human dose levels. In rats, acyclovir produced a nonsignificant increase in
chromosomal damage at 62 to 125 times human levels. No activity was observed
in a dominant lethal study in mice at 36 to 73
times human levels."
first paragraph, first sentence revised
"Acyclovir administered during organogenesis was not teratogenic in the mouse (450 mg/kg per day, PO), rabbit (50 mg/kg per day, SC and IV), or rat (50 mg/kg per day, SC)."
second paragraph, third sentence revised
"There were 749 756 pregnancies followed in women exposed to systemic acyclovir during the first trimester of pregnancy resulting in 756 outcomes."
Geriatric Use: Clinical studies of ZOVIRAX did not include sufficient numbers of patients aged 65 and over to determine whether they respond differently than younger patients. Other reported clinical experience has not identified differences in responses between elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased renal function, and of concomitant disease or other drug therapy."
Pediatric Use, "Safety and effectiveness in pediatric patients less than 2 years of age have not been adequately studied. of oral formulations of acyclovir in pediatric patients less than 2 years of age have not been established."
Geriatric Use: Of 376 subjects who received ZOVIRAX in a clinical study of herpes zoster treatment in immunocompetent subjects greater than or equal to 50 years of age, 244 were 65 and over while 111 were 75 and over. No overall differences in effectiveness for time to cessation of new lesion formation or time to healing were reported between geriatric subjects and younger adult subjects. The duration of pain after healing was longer in patients 65 and over. Nausea, vomiting, and dizziness were reported more frequently in elderly subjects. Elderly patients are more likely to have reduced renal function and require dose reduction. Elderly patients are also more likely to have renal or CNS adverse events. With respect to CNS adverse events observed during clinical practice, somnolence, hallucinations, confusion, and coma were reported more frequently in elderly patients (see CLINICAL PHARMACOLOGY, ADVERSE REACTIONS: Observed During Clinical Practice, and DOSAGE AND ADMINISTRATION)."
ADVERSE REACTIONS
Observed During Clinical Practice
General: Anaphylaxis, angioedema, …
Nervous: Aggressive behavior, agitation, ataxia, coma, confusion, decreased consciousness, delirium, dizziness, encephalopathy, hallucinations, paresthesia, psychosis, seizure, somnolence, tremors. These symptoms may be marked, particularly in older adults or in patients with renal impairment".
Hemic Hematologic and Lymphatic: Anemia, leukocytoclastic vasculitis, ….
Hepatobiliary Tract and Pancreas: Elevated liver function tests, hepatitis, hyperbilirubinemia, jaundice
OVERDOSAGE
second sentence revised
Overdoses involving ingestion of up to 100 capsules (20 g) have been reported.
Adverse events that have been reported only
association with overdosage include agitation, coma,
convulsions, seizures,
and lethargy.
Return to Product Menu | MedWatch Home | MedWatch Safety Info | Online MedWatch Report | Contact Medwatch
HTML by JLW