 |
 |
 |
|
 |
FDA Home Page | Search FDA Site | FDA A-Z Index | Contact FDA
Printable version (614 KB PDF)
July 16, 2008
Prepared by Booz Allen Hamilton Inc.
Prescription Drug User Fee Act III – Initiatives & Evaluations
Contract No. 223-04-8100 Task No. 3
TABLE OF CONTENTS
STUDY OVERVIEW
Objectives and Scope
Study Cohorts
Methodology
FINDINGS
Product and Disease Characteristics
GRMPs Compliance
Issues and Communication
Sponsor Characteristics
FDA Characteristics
APPENDIX A: GRMPS IMPLEMENTATION STATUS
GRMPs Compliance Baseline
GRMPs Compliance for FY 2005 to FY 2007 Products
APPENDIX B: SPONSOR AND FDA FOCUS GROUP FINDINGS
EXHIBITS
Exhibit 1. Study Objectives
Exhibit 2. Overview of Study Cohorts and Data Sources
Exhibit 3. Data Gathering and Analysis Methodology
Exhibit 4. Drivers and Hypotheses of Multi-Cycle Reviews
Exhibit 5. Overview of Data Sources
Exhibit 6. Product Application Cohorts, Data Sources, and Analyses
Exhibit 7. Cohort Product Status and Approval Rates
Exhibit 8. First Cycle Approval Rate for Overall Study Cohort by Year
Exhibit 9. Approval Rate vs. Novelty and Indication
Exhibit 10. Special Development and Review Programs
Exhibit 11. First-Cycle Approval Rate by Application Type
Exhibit 12. GRMPs Phases and Activities
Exhibit 13. First Cycle Approval Rate by GRMPs Compliance
Exhibit 14. First Cycle Approval Rate by GRMPs Compliance and Review Designation
Exhibit 15. GRMPs Compliance and FDA-Sponsor Communication Frequency and Early Communication Timing
Exhibit 16. Overview of FDA-Sponsor Communications
Exhibit 17. Effect of End of Phase 2 Meetings on Approval Rate for FY2005-FY2007 Cohort
Exhibit 18. Effect of Pre-NDA/BLA Meetings and Timing on Approval Rate for FY2005-FY2007 Cohort
Exhibit 19. Incidence of Pre-NDA/BLA and EOP2 Meetings
Exhibit 20. Filing Review Issues Identified by Year
Exhibit 21. Impact of Issue Identification in Filing Review Notification for FY2002-FY2007 Cohort
Exhibit 22. Applications with All Filing Review Issues Resolved by Action Date for FY2005-FY2007 Cohort
Exhibit 23. Approvability Impact of 74-Day Letter Issues
Exhibit 24. FDA-Sponsor Communications for FY2005-FY2007 Cohort
Exhibit 25. Communication Type for FY2005-FY2007 Cohort
Exhibit 26. Amendments Submitted for Applications for FY2005 Products
Exhibit 27. Types of Amendments Submitted for FY2005 Applications
Exhibit 28. Advisory Committee Meetings and Review Designation
Exhibit 29. Advisory Committee Meeting Timing and Priority/Standard Designation
Exhibit 30. Impact of Major Issues Identified on Approval Rate in FY2005-FY2007 Cohort
Exhibit 31. Major Deficiencies Cited in First Action Letter of Multi-Cycle Applications by Category for FY2002-FY2007 Cohort
Exhibit 32. Major Deficiencies Cited in First-Cycle Action Letter of Multi-Cycle Applications by Area for FY2002-FY2007 Cohort
Exhibit 33. Examples of Issues Observed by Category
Exhibit 34. Products with Postmarketing Study Commitments in FY2002-FY2007 Cohort
Exhibit 35. Distribution of PMCs for Single- and Multi-Cycle Approvals
Exhibit 36. Focus Area of Postmarketing Study Commitments for FY2002-FY2007 Cohort
Exhibit 37. Single-Cycle Approval Rate by Sponsor Experience
Exhibit 38. Sponsor Experience and Impact of Pre-Submission Meetings
Exhibit 39. Cumulative Average First Cycle Communications During Review Period by Sponsor Experience
Exhibit 40. Approval Rate vs. Sponsor Type and Origin
Exhibit 41. First Cycle Approval Rate by Review Designation and Company Size
Exhibit 42. Deficiencies in Multi-Cycle Products by Sponsor Size in FY2005-2007
Exhibit 43. Cumulative Average First-Cycle Communications During Review Period by Company Size
Exhibit 44. Cohort Applications by Office and Review Designation
Exhibit 45. Submission Timing and First Cycle Approval Rates
Exhibit 46. Staffing Changes Between Pre-Submission and Review
Exhibit 47. Impact of Foreign or Domestic Manufacturing Site
Exhibit 48. Summary Overview of Recommendations
Exhibit 49. Baseline of GRMPs Practices Adopted by Divisions – January 2006 Snapshot
Exhibit 50. GRMPs Compliance by Product
Exhibit 51. CDER and CBER GRMPs Compliance by Office
Exhibit 52. GRMPs Compliance in Review Phase
Exhibit 53. GRMPs Compliance in Filing and Action Phases
Exhibit 54. Review Communication Practices Preferred by Industry and FDA Focus Group Participants
Exhibit 55. Industry and FDA Focus Group Feedback on GRMPs Filing and Planning and Review Activities
Exhibit 56. Industry and FDA Focus Group Feedback on GRMPs Advisory Committee, Action and Post-Action Phases
In conjunction with the 2002 Prescription Drug User Fee Act (PDUFA) Reauthorization Performance Goals and Procedures (PDUFA III Goals), the Food and Drug Administration (FDA) agreed to meet specific performance goals to improve the effectiveness and efficiency of FDA review of New Drug Applications (NDAs) and Biologic License Applications (BLAs). Several of these goals are focused on improving the review process activities occurring between initial submission of the application and subsequent FDA action regarding the application (i.e., first-cycle review). The focus of this study is to identify and examine what factors contribute to and detract from FDA’s ability to make an approval decision during the first-cycle review for products that are ultimately approved without major new data submissions. This evaluation contract was to determine the impact of FDA’s initiatives to enhance first-cycle review performance during the five-year period of PDUFA III. The scientific merit of an application, while of critical importance to approval decisions, was not within the scope of this study; nonetheless, a significantly deficient application cannot be expected to attain first-cycle approval, even if the product is ultimately found to be safe and effective.
Booz Allen conducted a two-part study of first-cycle review initiatives: a Retrospective Analysis[1] and a Prospective Analysis. The studies focused on the first-cycle review processes that are conducted on NDAs for new molecular entities (NMEs) and BLAs. The Overall Study Cohort included 185 NME NDAs and BLAs that were received during fiscal years 2002-2007 and reached first action by September 30, 2007. The first-cycle approval rate for the 185 applications was 50%. The FY2002 – FY2007 cohort was comprised of 74% NDAs and 26% BLAs. The NDAs had a 43% first-cycle approval rate, while the BLAs rate was higher for both Center for Drug Evaluation and Research (CDER) (86%) and Center for Biologics Evaluation and Research (CBER) (66%).
The study examined several product and disease characteristics, including review designation, condition severity and mechanism of action. Priority review designation, which is given to applications for products that offer major advances in treatment or provide a treatment where no adequate therapy exists, had the most significant impact on first-cycle approval rates. Within this FY2002 – FY2007 cohort, 45% had Priority designation. Applications with a Priority designation had a higher first-cycle approval rate (68%) than products with Standard review designation (36%) (Note that Priority reviews have a six month goal for initial action, while Standard reviews have a ten month goal for initial action). Booz Allen speculates that this higher first-cycle approval rate for Priority approval drugs reflects a greater effort to resolve issues during the first cycle, which is consistent with FDA’s commitment to provide access to therapies for unmet medical needs. Products with a novel mechanism of action (MOA) targeting life-threatening conditions had a greater first-cycle approval rate (62%) compared to that of products with non-novel MOAs addressing non-life-threatening conditions (39%).
We observed that applications that complied with most or all of the assessed Good Review Management Principles and Practices (GRMPs) activities had the highest first-cycle approval rates. For applications assessed after the FY2005 GRMPs rollout, application reviews that complied with 80% of assessed GRMPs activities and timeframes [2] or more had a first-cycle approval rate of 71% compared to the first-cycle approval rate of 50% for those application reviews that complied with 20% of GRMPs assessed activities. In conducting a closer examination of the assessed GRMPs activities and timeframes, there was no single factor that contributed to the higher first-cycle approval rate.
Recommendation:
Booz Allen recommends that FDA continue with GRMPs implementation, ensuring adoption of both GRMPs activities and timeframes. Also, Booz Allen recommends a quality system approach for process improvement and feedback through evaluating quantitative metrics and staff perceptions.
The resolution of major application issues was critical to first-cycle approval. In many instances issues identified early in the review process by FDA were not addressed by the sponsor in a timely manner or were not resolved during the first cycle. Applications with major deficiencies identified and documented by FDA either pre-submission or during the review were less likely to be approved in a single cycle than those applications that did not have a major deficiency identified during the same timeframe. However, applications were more likely to be approved in the first cycle if a major deficiency was identified pre-submission (40%) than if major deficiencies were identified during the review (19%). Applications for which no major deficiency was identified either pre-submission or during the review had a high first-cycle approval rate (92%). The majority of multi-cycle applications have major deficiencies in one or two key categories; these were in the critical areas of safety, efficacy or chemistry manufacturing and controls (CMC).
Sponsors continue to take advantage of the pre-submission meeting opportunities: End of Phase 2 (EOP2) and Pre-NDA/BLA. There was a significant increase in the number of applications that had an EOP2 and/or Pre-NDA/BLA meeting from FY2002 to FY2007. In the Prospective Analysis Cohort, 78% of sponsors participated in EOP2 meetings and 93% participated in Pre-NDA/BLA meetings. In addition, sponsors who submitted their application to FDA within six months of the Pre-NDA/BLA meeting had a 71% first-cycle approval rate, as compared to a 39% first-cycle approval rate for those that submitted their application more than six months from the date of the meeting.
The Filing Review Notification, or 74-Day Letter, is an effective tool in communicating deficiencies to sponsors. For applications submitted FY2005 – FY2007, 62% of these applications had all potential review issues [3] that were listed in the 74-Day Letter resolved by the action date. Of those that resolved the potential review issues conveyed in the letter, 62% were approved in the first cycle, indicating that FDA successfully identified and communicated important review issues to the sponsor in the Filing Review Notification.
Throughout the review cycle, the FDA and sponsors frequently engage in informal communications (email, telephone, and fax) in addition to face-to-face meetings. Analysis of Action Packages [4] and FDA systems demonstrated that the frequency of FDA-sponsor communications was similar for single-cycle and multi-cycle reviews for the first three-quarters of the application review cycle. In the final quarter of the review cycle, however there was a significant increase in communications for single cycle approvals compared to multiple cycle applications. Approximately 70% of these additional communications were related to labeling and postmarketing study commitments (PMC) issues, which is not surprising since these communications are needed in the final stages of an approval action.
Recommendation:
Booz Allen recommends a continued shift in emphasis of discussing data in addition to application format issues in key pre-submission meetings so as to promote earlier issue identification and resolution. Booz Allen also recommends that FDA should continue to use the 74-Day Letter to communicate application deficiencies early in the review process. To improve the efficiency and the effectiveness of the review process, Booz Allen recommends that FDA and sponsors agree on an application review communication protocol during filing determination, which would set expectations for sponsors about appropriate methods (e.g., secure email) and timeframes for FDA communication.
Sponsor size was a contributing factor to first-cycle approval rates, with large companies receiving first-cycle approval 58% of the time, compared to only 41% for small companies. Large companies were more likely to have prior FDA experience than small companies, a factor which is also correlated with higher first-cycle approval rates. These larger, more experienced companies often took advantage of opportunities to discuss product development issues in pre-submission meetings than smaller companies. Further, large companies on average had more communications with FDA throughout the application review cycle than smaller companies, which may have helped them to more rapidly identify and address potential review issues. Even for multi-cycle applications, smaller sponsors tended to have deficiencies covering a wider spectrum included in the action letter compared to larger sponsors, further suggesting that larger companies have fewer review issues and are able to leverage their experience and resources to identify and resolve issues in a timely manner. Booz Allen speculates inexperienced sponsors would probably benefit from additional FDA-initiated efforts to clarify review processes and guidances on product development.
Recommendation:
Booz Allen recommends that sponsors should maximize the efficiency and the effectiveness of the review process by tracking and resolving the issues identified during pre-submission meetings (e.g., EOP2, Pre-NDA/BLA), and actively resolving those issues identified during the review phase to the extent practicable.
The FDA received a significantly higher number of NDAs for NMEs and BLA submissions in the fourth quarter (44% of annual total) compared to any other quarter in the calendar year (19% of total per quarter, on average). These applications had the lowest rate of first-cycle approvals. Examination of other factors (e.g., review designation, sponsor size, sponsor experience) did not provide any insight regarding the significant differences with these applications in comparison to applications submitted in other quarters. Booz Allen speculates that application quality may contribute to the lower first-cycle approval rate of fourth quarter submissions; however, the quality of an application was not within the scope of this study.
PDUFA, enacted in 1992 and renewed in 1997 (PDUFA II), 2002 (PDUFA III), and 2007 (PDUFA IV), authorizes FDA to collect fees from companies that produce certain human drug and biological products. These revenues provide FDA with additional resources to expedite and improve the review of human drug and biologic product applications. In conjunction with the PDUFA III Goals, FDA agreed to meet specific performance goals to improve the effectiveness and efficiency of FDA review of NDAs and BLAs. [5] Several of these goals are focused on improving the review process activities occurring between initial submission of the application and subsequent FDA action regarding the application (i.e., first-cycle review). The focus of this study is to identify and examine factors, excluding scientific merit, that contribute to and detract from FDA’s ability to approve an application during the first-cycle review.
FDA, industry and the public benefit when a well-managed application review process allows sponsors with applications that meet the safety and efficacy standards for approval to resolve all issues (e.g., clarification of, or additional analyses, negotiation of labeling, and postmarketing study commitments) within a single review cycle. Efficient review allows FDA to fulfill its public health mission to ensure that safe and effective products are available to the public in a timely manner and allows for efficient use of resources. The public benefit from timely access to safe and effective therapies and first cycle approval allows the sponsoring company to market the product sooner and capture revenues. Yet, not all products should or can be approved in a single cycle. If FDA uncovers substantial deficiencies during an application’s first-cycle review or a sponsor does not respond to requests for information in a timely manner, then additional review cycles may be necessary to address all deficiencies in an application.
The PDUFA goals specify that FDA will retain an independent expert consultant to evaluate the application review process improvement initiatives and the impact of GRMPs. FDA has contracted Booz Allen to perform the independent program evaluation of the application review process.
The goal of the evaluation was to identify factors that contributed to or detracted from first-cycle Approvals, as well as determine the impact of FDA’s implementation of two initiatives to enhance first-cycle review processes and performance during the five-year period of PDUFA III. In the first initiative, CDER and CBER created a joint guidance, Good Review Management Principles and Practices (GRMPs), which describes a well-managed process for review of NDAs, BLAs and efficacy supplements, particularly during the first-cycle review. One of the goals of the GRMPs was to improve the rate of first cycle approvals without altering the approval standards. FDA believes that it is in everyone’s best interest , FDA, the sponsor, and society if an otherwise approvable drug is approved during the first cycle. For the second initiative, FDA agreed to provide applicants with early notification of potential NDA and BLA review issues identified during the filing review (i.e., the 74-Day letter). The scientific merit of an application, while of critical importance to the regulatory action, was beyond the scope of this study.
Booz Allen conducted a two-part study of first-cycle review initiatives: a Retrospective Analysis and a Prospective Analysis. The studies focused on the first-cycle review processes for marketing applications for NMEs and BLAs. The Retrospective Analysis, published in January 2006 [6], represented an interim analysis of first-cycle approval factors for NME applications submitted between FY2002 and FY2004. The Prospective Analysis analyzed the remaining PDUFA III applications. Specific objectives of the two Analyses are discussed in Exhibit 1.
Exhibit 1. Study Objectives
| Study | Objectives |
|---|---|
| Retrospective Analysis |
|
| Prospective Analysis |
|
This report reflects integrated findings from the entire first-cycle review initiative evaluation study (Retrospective and Prospective Analyses). This report also includes an assessment of GRMPs impact (Appendix A) and the sponsor perspective gained through interviews and focus groups (Appendix B).
The total product applications evaluated for this study included 185 NME NDAs and BLAs that were received during fiscal years 2002-2007 and reached first action by September 30, 2007 [7]. The 185 products were divided into two cohorts, the Retrospective Analysis and Prospective Analysis.
For the Retrospective Cohort, Booz Allen’s primary source of information was FDA documented summaries of the application review outcomes, commonly referred to as Action Packages. For the Prospective Analysis Cohort, Booz Allen supplemented this information with additional FDA data, as well as solicited sponsor perspectives. A summary of the timeframes and data associated with each cohort is presented in Exhibit 2.
Exhibit 2. Overview of Study Cohorts and Data Sources

A more detailed discussion of our approach and sources for data collection is discussed in the following section.
Booz Allen followed a systematic methodology for conducti both the Retrospective and Prospective Analyses (Exhibit 3). The predominant difference between the Retrospective and Prospective Analyses were the data sources available for analysis.
Exhibit 3. Data Gathering and Analysis Methodology
| 1. Identify Potential Drivers of Multi-Cycle Review | 2. Gather Data | 3. Analyze Data and Test Hypotheses | 4. Develop Findings & Recommendations |
|---|---|---|---|
|
Leverage FDA Documents and Systems
|
|
|
Note: Asterisk(*) denotes activities performed only in the Prospective Analysis
The following sections provide additional details of our four step methodology, with particular focus on the specific factors analyzed and the data sources used to support the analysis.
The first step consisted of generating hypotheses of potential drivers of single/multiple review cycles and identifying the indicators and/or metrics appropriate for hypothesis testing (Appendix C contains a complete list of hypotheses generated). Booz Allen generated the initial study hypotheses based on our knowledge of the FDA review process and industry experience with the process, as well as consultation with FDA leadership. These hypotheses evolved throughout the duration of the study based on observations, additional analysis, as well as feedback from industry focus groups and FDA leadership. In addition, there were several hypotheses (e.g., application quality, reviewer/division workload) that were not evaluated based on the absence of appropriate indicators or metrics. The hypotheses can be categorized in five major categories: Product and Disease Characteristics, GRMPs Compliance, Issues and Communication, Sponsor Characteristics and FDA Characteristics (Exhibit 4). The full list of hypotheses identified is included in Appendix C: Study Hypotheses.
Exhibit 4. Drivers and Hypotheses of Multi-Cycle Reviews
| Factors | Key Analysis Areas |  |
Sample Hypotheses |
|---|---|---|---|
 |
|
|
Note: Red Italics indicate hypothesis categories that were not tested because appropriate test indicators or metrics could not be identified or insufficient data existed.
In conducting this study, Booz Allen used multiple sources of data: FDA systems, Action Packages, review observation during FDA internal meetings, sponsor interviews, FDA focus groups and sponsor focus groups. Specific data sources are summarized in Exhibit 5.
Exhibit 5. Overview of Data Sources
| Data Source | Description of Data Source and Use |
|---|---|
| FDA Action Packages | A primary source of data for both the Retrospective and Prospective Analyses was FDA-compiled product Action Packages which contain records of FDA internal and FDA-sponsor communications and application review documents. |
| Systems that Support Application Review | During the Prospective Analysis, FDA systems that support NDA and BLA application review were used as a supplementary source of information on review communications, timelines, and issues. |
| Observation of Application Review Meetings | Since data available in FDA documents and systems were insufficient to test some hypotheses, additional data were gathered by observing the specific aspects of the application review process during the Prospective Analysis. Booz Allen observers attended review meetings (e.g., mid-cycle meetings, pre-approval safety conferences, and labeling meetings) that represented a broad spectrum of activities, including milestones as specified in GRMPs. Multiple divisions and offices were represented in the observed meetings. Beginning mid-2006, forty-one application reviews were observed for this activity. |
| Review Team Input | Where feasible and not available through other sources, input from FDA Regulatory Project Managers and other review team members was solicited regarding: application review activities, FDA-sponsor interactions (format, frequency and timing), nature and timing of issues identified. |
| Sponsor Interviews | Booz Allen conducted 20 sponsor interviews post-action, to gather data and industry perspectives on the FDA review processes. Interviews were held post-action to ensure the evaluation activities did not disrupt or potentially influence the review. Sponsors with applications submitted during FY2006 and FY2007, which reached action by June 2007 (33 products) were invited to participate in a post-action interview with Booz Allen. Of the invitees, 20 sponsors accepted and were interviewed; these included: 7 large pharmaceutical companies (35%), 3 medium pharmaceutical companies (15%), 5 small pharmaceutical companies (25%), 2 large biotechnology companies (10%), 2 small biotechnology companies (10%), and 1 research institution (5%). Interview discussions focused on FDA-sponsor communication, GRMPs impact, and specific application experiences. |
| Industry Focus Groups | Booz Allen conducted two, one day focus group meetings with industry to gather perceptions of the GRMPs and perspectives on the root causes for multi-cycle review. Communication effectiveness between sponsors and FDA was also discussed. |
| FDA Focus Groups | Booz Allen conducted two, half-day focus group meetings with FDA review divisions to gather perspectives on GRMPs implementation status, challenges with implementation, effectiveness of GRMPs training, and perspectives on root causes of multi-cycle review. |
| Publicly Available Data Sources | Data sources such as company websites were used to supplement product and sponsor company background information such as product/disease characteristics and sponsor profiles (e.g., company size, previous experiences with FDA). |
Data were analyzed using both quantitative and qualitative methods to test hypotheses developed. However, the small numbers of product applications limited the ability to demonstrate statistically significant analyses. In addition, the number of product applications meeting the test criteria was even further limited (for example, novel mechanism of action coupled with product origin: in-house vs. acquired technology). As such, most quantitative analyses were limited to basic statistics (e.g., mean, frequency, range). Qualitative analyses were used to assess factors such as GRMPs impact and effectiveness of FDA/Sponsor communications. Where possible and practical, the study draws logical inferences based on Booz Allen observations and discussions with FDA, however, the small number of product applications (and subsets of applications) impacted the ability to generalize conclusions in some instances. In the process of testing hypothesis through data collection and analysis, we eliminated some hypothesis based on inadequate data quality or insufficient data.
While many hypotheses were identified, only those hypotheses that had notable findings were retained. Using both qualitative and quantitative analyses, Booz Allen generated findings consistent with the study objectives, focusing on the following areas:
Based on these findings, Booz Allen developed recommendations focused on activities that FDA and/or industry could perform to address our findings, particularly to improve first review cycle outcomes. Recommendations were categorized based on the review phases (i.e., pre-submission, filing and planning, review, Advisory Committee, action, post-action).
The following sections present the analysis and implications from the five-year study of factors that contribute to or detract from FDA’s ability to make a decision regarding the product application on first-cycle. Major findings are described by hypothesis categories: Product and Disease Characteristics, GRMPs Compliance, Issues and Communication, Sponsor Characteristics and FDA Characteristics. In most instances, our findings are representative of the Overall Study Cohort. However, when there was a notable difference between the findings from the FY2002– FY2004 and FY2005 – FY2007 cohorts, we present these findings by cohort. In addition, there are some analyses that were conducted using a subset of the Overall Study Cohort, based on availability and quality of data (Exhibit 6).
Exhibit 6. Product Application Cohorts, Data Sources, and Analyses [8]
| Cohort | Number of Applications | Analyses | Data Collection Method | ||
|---|---|---|---|---|---|
| Action Packages | Public Data Sources | FDA Application Review Systems | |||
| Overall Study Cohort | 185 |
|
 |
|
|
| Prospective Analysis Cohort | 85 |
|
|
|
|
The quantitative and qualitative data collected through observation of application review team meetings, review team input, sponsor interviews, industry focus groups, and FDA focus groups were used in the development of findings and recommendations. Detailed findings for the five categories of hypotheses tested are listed in the following sections.
Exhibit 7. Cohort Product Status and Approval Rates
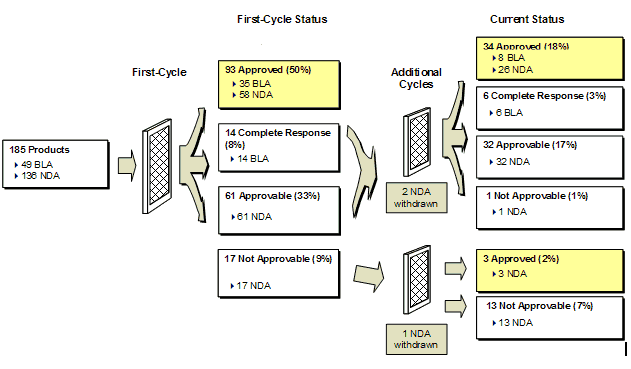
The first-cycle approval rate for individual years throughout the FY2002 - FY2007 cohort ranged from 45% to 53%, with the overall Retrospective Analysis Cohort having a first-cycle approval rate (49%) similar to the Prospective Analysis Cohort (52%).
The FY2002 - FY2007 cohort was comprised of 74% NDAs and 26% BLAs (Exhibit 8). The NDAs had a 43% first-cycle approval rate, while the BLAs rate was higher for both CDER (86%) and CBER (66%). Of the FY2002 - FY2007 cohort applications, products with Priority designation (45%) had a higher approval rate (68%) than products with Standard designation (36%).
Exhibit 8. First Cycle Approval Rate for Overall Study Cohort by Year
| Fiscal Year | |||||||
|---|---|---|---|---|---|---|---|
| 2002 | 2003 | 2004 | 2005 | 2006 | 2007 | Overall | |
| Total Product Applications | 29 | 35 | 36 | 38 | 32 | 15 | 185 |
| Approval Rate | 45% | 51% | 50% | 50% | 53% | 53% | 50% |
| Application Type | |||||||
| NDAs | 21 | 29 | 28 | 29 | 21 | 8 | 136 |
| Approval Rate | 52% | 45% | 36% | 41% | 43% | 38% | 43% |
| BLAs | 8 | 6 | 8 | 9 | 11 | 7 | 49 |
| Approval Rate | 25% | 83% | 100% | 78% | 73% | 71% | 71% |
| Review Designation | |||||||
| Standard | 20 | 19 | 18 | 17 | 19 | 9 | 102 |
| Approval Rate | 35% | 42% | 39% | 24% | 37% | 44% | 36% |
| Priority | 9 | 16 | 18 | 21 | 13 | 6 | 83 |
| Approval Rate | 67% | 63% | 61% | 71% | 77% | 67% | 67% |
| Application Type and Review Designation | |||||||
| NDA | |||||||
| Standard | 13 | 17 | 13 | 14 | 15 | 6 | 78 |
| Approval Rate | 38% | 35% | 15% | 14% | 33% | 33% | 28% |
| Priority | 8 | 12 | 15 | 15 | 6 | 2 | 58 |
| Approval Rate | 75% | 58% | 53% | 67% | 67% | 50% | 62% |
| BLA | |||||||
| Standard | 7 | 2 | 5 | 3 | 4 | 3 | 24 |
| Approval Rate | 29% | 100% | 100% | 67% | 50% | 67% | 63% |
| Priority | 1 | 4 | 3 | 6 | 7 | 4 | 25 |
| Approval Rate | 0% | 75% | 100% | 83% | 86% | 75% | 80% |
The higher first-cycle approval rate for Priority-designated drugs, suggesting greater effort to resolve outstanding issues during that cycle, is consistent with FDA’s commitment to provide access to therapies for unmet medical needs. Given the pronounced effect of review designation (e.g., Priority vs. Standard), Booz Allen reviewed the impact of the review designation on all analyses presented. When the review designation resulted in a significant finding for the analyses, the data were presented.
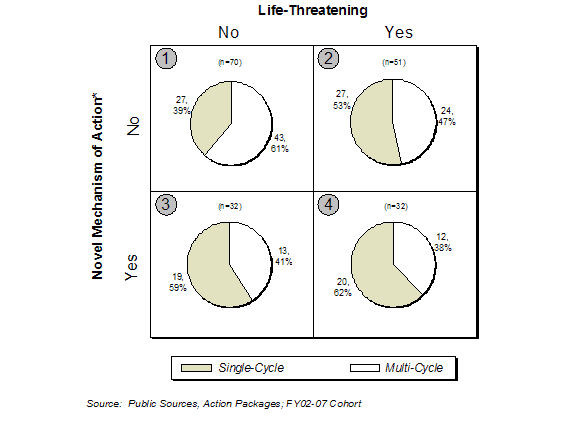
In Exhibit 9, products with a novel mechanism of action and indication for a life-threatening condition had a slightly higher rate of first-cycle approval (62%) compared to products that met none of these criteria. Product applications for life threatening indications had a first-cycle approval rate of 53% and novel mechanism of action applications had a first-cycle approval rate of 59%. Non-novel products for non-life threatening conditions had a first-cycle approval rate of 39%. Booz Allen speculates the severity of the medical conditions addressed, the different levels of acceptable risk, and the urgency for new products might explain these findings.
Exhibit 9. Approval Rate vs. Novelty and Indication
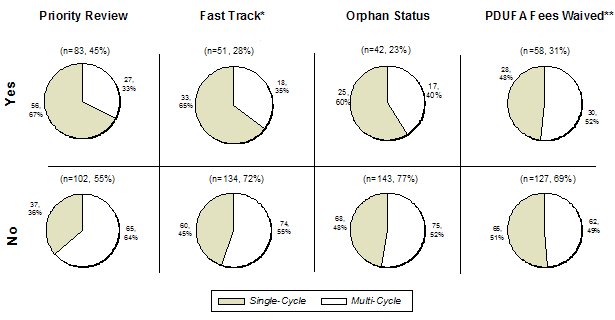
FDA has developed several programs to expedite review and facilitate product development (Exhibit 10). Fast Track and Priority review programs are used to facilitate the sponsor-FDA interactions and expedite the review processes for products that address serious diseases with significant unmet medical needs. For products that qualify for Fast Track, which is granted during the Investigational New Drug (IND) phase, the FDA may engage in more pre-submission meetings and communications with the sponsors, as well as consider reviewing portions of the application submitted prior to complete submission of the NDA/BLA. Many Fast Track-designated products also qualify for Priority review, which is granted after the NDA/BLA is submitted, and sets the target date to complete application review at six months (standard reviews have a ten-month target date for completion). Orphan product status, as well as other fee waiver opportunities (e.g. small business waiver), provide financial incentives to sponsors but do not impact the review process independently of a Priority review designation.
Exhibit 10. Special Development and Review Programs
| Fast Track | Priority Review | Orphan Status | Fee Waivers | |
|---|---|---|---|---|
| Program Benefits |
|
|
|
|
| Eligibility Requirements |
|
|
|
|
These expedited development and review programs seemed effective in driving single-cycle approvals, as 67% of applications with Priority reviews received first-cycle approval, compared to only 36% for non-Priority applications (Exhibit 11). Similarly, higher first-cycle approval rates were observed for Fast Track and Orphan product applications. Qualification for a fee waiver, however, did not impact first-cycle outcomes, with similar approval rates for applications with or without the fee waiver (48% vs. 51%, respectively). Notably, many Orphan product applications also received Fast Track and/or Priority review designation. Out of the 51 products with Fast Track designation, 32 of the products were submitted by large companies, and 27 (84%) of these products were approved in a single review cycle. Of the Priority review applications, 29 also had Orphan designation, with 21 (72%) of these applications achieving first-cycle approval. Of the 54 remaining Priority review applications, 35 (65%) achieved first-cycle approval. A lack of sponsor’s regulatory experience may be a compounding factor in the first-cycle approval rates for the applications that qualified for a fee waiver since most of these products were developed by small companies with no previously approved products (see Sponsor Characteristics).
Exhibit 11. First-Cycle Approval Rate by Application Type
Several additional product and disease characteristics were analyzed in the study, but did not have an impact on first-cycle approval rates. There was no significant difference in approval rates for products addressing chronic or acute conditions. Similarly, differences in product origin (in-house or in-licensed) did not have any effect on the approval rate in the first cycle. Finally, products with data from international clinical sites or that had prior foreign regulatory approval had similar first-cycle approval rates as those products that did not.
In April 2005, FDA formalized GRMPs through the issuance of Guidance for Review Staff and Industry – Good Review Management Principles and Practices for PDUFA Products as agreed upon in the PDUFA III goals. This guidance is intended for industry and review staff and focuses on good review management principles and practices as they apply to the first-cycle review of NDAs, BLAs, and efficacy supplements. The GRMPs guidance is based on the collective experience of CDER and CBER with review of applications for PDUFA products. The guidance is intended to promote the practice of good review management based on sound fundamental values and principles. Adoption of GRMPs is intended to improve the quality, efficiency, transparency and consistency of the application review process. [10] The draft GRMPs guidance was published June 28, 2003, and the final guidance in April 2005.
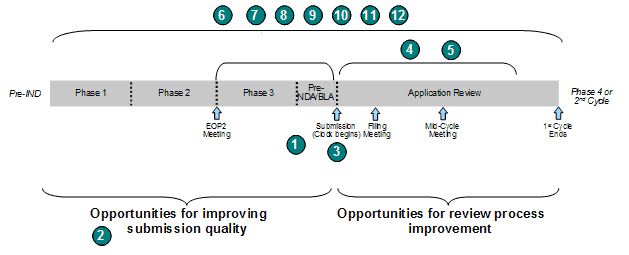
The GRMPs divide the NDA/BLA review process into five distinct phases: filing determination and review planning, review, Advisory Committee meeting preparation and conduct, action, and post-action. Within these phases, the guidance further identifies 42 review activities and associated timelines for completion within the phases of the review (Exhibit 12).
Exhibit 12. GRMPs Phases and Activities
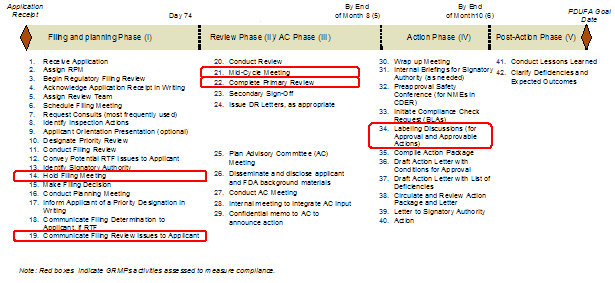
In 2006, Booz Allen analyzed the status of GRMPs implementation, the impact of GRMPs, and the sponsor perception of GRMPs on the review process (Appendix A: GRMPs Implementation Status, Appendix B: Sponsor and FDA Focus Group Findings). For the FY2005 - FY2007 cohort, Booz Allen evaluated GRMPs compliance and impact by examining five key activities and their associated timelines as they applied to product application reviews:
The choice of these five activities was based on the importance of the activity, as well as the availability of information to assess compliance with these activities. Specifically, these activities provided consistent documentation (e.g., meeting minutes, correspondence) to determine the timing and existence of these activities. In determining compliance rates, Booz Allen assessed whether the activity was conducted within the timeframes. For example, if a review team conducted three of the five assessed activities within the prescribed GRMPs timeframes, the compliance rate was captured as 60% (3 of 5 tasks).
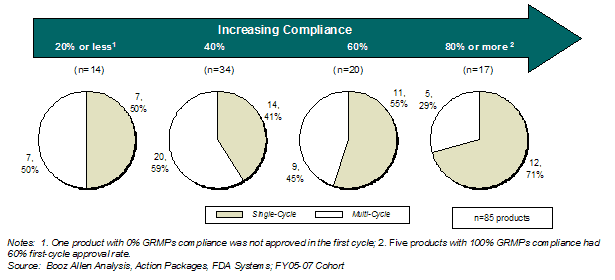
We observed that applications that complied with most or all of the assessed GRMPs activities had the highest first-cycle approval rates (Exhibit 13).
Exhibit 13. First Cycle Approval Rate by GRMPs Compliance
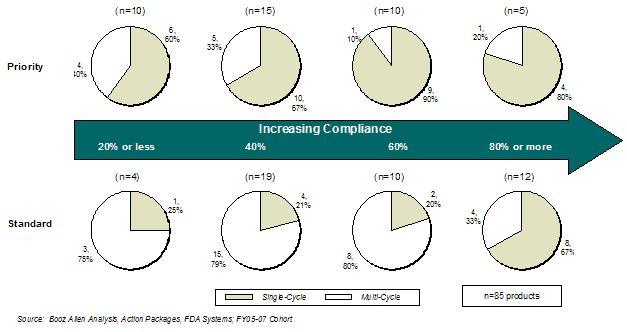
For the standard review products that had 20%, 40%, or 60% compliance, the overall first-cycle approval rate was similar (Exhibit 14).
Exhibit 14. First Cycle Approval Rate by GRMPs Compliance and Review Designation
Booz Allen further examined first-cycle approval rates associated with increased GRMPs compliance to identify other factors (e.g., early communication, communication frequency) that may be related to or responsible for higher first-cycle approval rates. However, the improved rates associated with GRMPs compliance could not be attributed to any specific aspect of GRMPs. With the limited data, Booz Allen did not perform a multi-variable analysis of GRMPs and other non-GRMPs factors that might affect first-cycle approval rates (e.g., application quality).
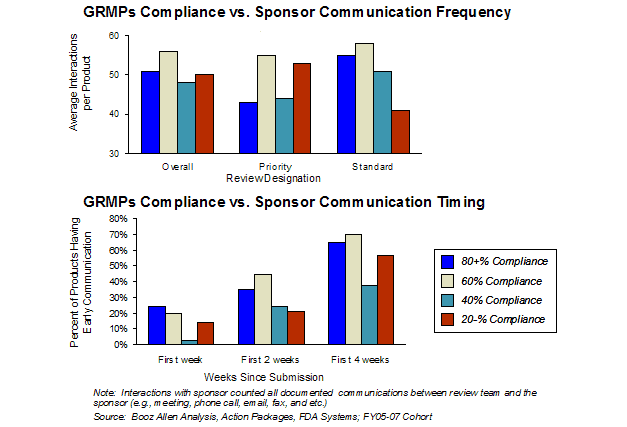
Compliance with assessed activities was not associated with earlier FDA communication with sponsors (Exhibit 15). Overall, GRMPs compliance had no association with the frequency of communications between the FDA and the sponsor during a product review. However, for Standard review designated applications, greater GRMPs compliance may have been associated with a slight increase in communication frequency.
Exhibit 15. GRMPs Compliance and FDA-Sponsor Communication Frequency and Early Communication Timing
While observing internal review meetings for the 41 FY2006-FY2007 products Booz Allen noted that review teams that captured post-meeting action items tended to spend less time revisiting issues in subsequent meetings, especially when action was needed by a discipline reviewer that did not participate full-time in the application review. While the observed sample size was too small to generalize, the structured meeting and documentation practice appeared beneficial. These observed activities were not required by GRMPs. The activities included scheduling regular team meetings, briefly summarizing team meeting minutes and action items, and consistently documenting information requests sent to sponsors.
Application issue identification and communication were key factors analyzed given the existence and severity of unresolved issues that result in multi-cycle reviews. Booz Allen examined interactions between FDA and sponsors prior to application submission and then during the review phase (Exhibit 16). Specific variables evaluated included the timing and frequency of meetings, timing of communication of application issues, and use of postmarketing commitments. Our analysis was limited to those communications and issues that were documented, reported, or observed. For pre-submission meetings, documentation was not available for all pre-submission events, but most significant milestone meetings (e.g., End of Phase 2) were included in the Action Packages or FDA systems, and comprised the basis for the pre-submission analyses. For review communications, we relied on Action Packages and FDA systems in combination with observations at review meetings and review team input.
Exhibit 16. Overview of FDA-Sponsor Communications
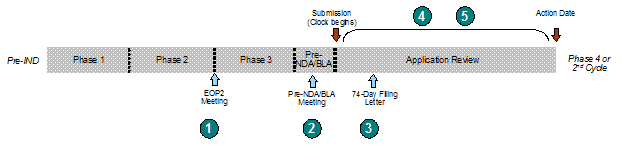
| Type of Communication | Timing | Expected Outcomes | |
|---|---|---|---|
| 1 | End-of-Phase 2 Meeting |
|
|
| 2 | Pre-NDA/BLA Meeting |
|
|
| 3 | Filing Review Issues (74-Day Filing) Letter |
|
|
| 4 | Information Requests (e.g., letter, fax, email, phone) |
|
|
| 5 | Amendments |
|
|
Pre-submission meetings between the FDA and sponsors provide an opportunity to review and discuss the status of the product in development and to agree on the planning for the subsequent stages of product testing and filing. The two main meetings that occur prior to application submission are the End of Phase 2 meeting(s) and Pre-NDA/BLA meeting. End of Phase 2 meetings are held after Phase 1 and Phase 2 clinical trials have been completed. These meetings are intended to determine whether a product is safe to proceed to Phase 3 clinical efficacy trials and agree on pivotal trial design. Pre-NDA/BLA meetings are conducted after the completion of all clinical studies that will be included in the submission.
Exhibit 17. Effect of End of Phase 2 Meetings on Approval Rate for FY2005-FY2007 Cohort
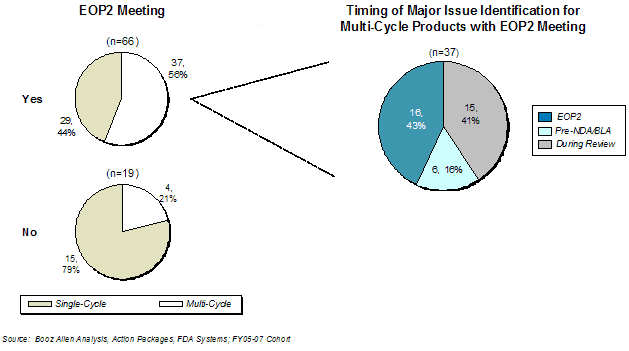
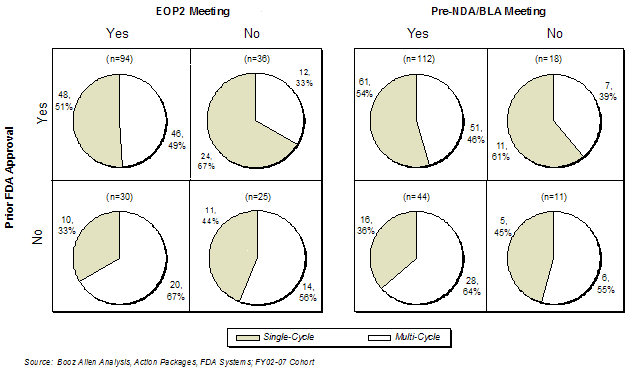
In the FY2005 - FY2007 cohort,[11] EOP2 meetings, perhaps surprisingly, did not appear to have a positive impact on first-cycle approval rate. Of the 66 products with EOP2 meetings, 44% received first-cycle approval, while 79% of products that did not have EOP2 meetings were approved in the first cycle (Exhibit 17). Of the 37 multi-cycle application reviews that had EOP2 meetings, the deficiency that led to the failure to achieve approval was identified in the EOP2 meeting 43% of the time. However, this observation stands in contrast to the data from the FY2002- FY2004 cohort, in which 52% of the 46 products that had an EOP2 meeting were approved in the first cycle vs. 29% for the 21 products that did not. Booz Allen notes that there was a significant increase in the number of applications that had EOP2 (78%) or Pre-NDA/BLA meetings (93%) in the FY2005 - FY2007 cohort compared to the FY2002 - FY2004 cohort.
The main purposes of the Pre-NDA/BLA meeting are to discuss the efficacy evidence from the Phase 3 trials, identify unresolved issues and agree on the format for the submission, including data presentation methods. In the FY2005-FY2007 cohort, Pre-NDA/BLA meetings were held for virtually all applications, so the approval rate nearly matches that of all products in the Overall Study Cohort (Exhibit 18).
Exhibit18. Effect of Pre-NDA/BLA Meetings and Timing on Approval Rate for FY2005-FY2007 Cohort
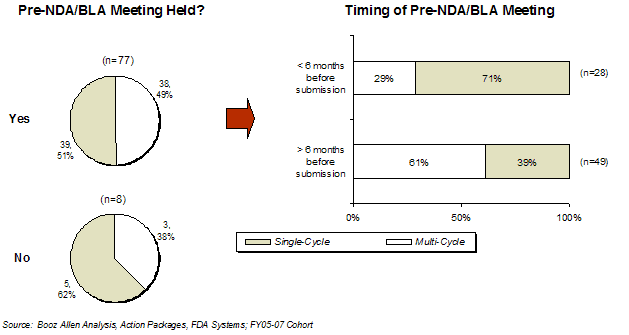
There appeared to be a difference observed in the first-cycle approval rate for products that submitted their application within six months of the Pre-NDA/BLA (71%) vs. those that waited more than six months (39%). Additionally, this higher first-cycle approval rate (71%) for applications submitted within six months was in contrast to the data in the FY2002- FY2004 cohort, in which only 46% were approved in the first cycle. Based on interviews with sponsors, Booz Allen speculates this improvement may be explained by a shift in emphasis by FDA on the data review, rather than application format issues, during the Pre-NDA/BLA meetings.
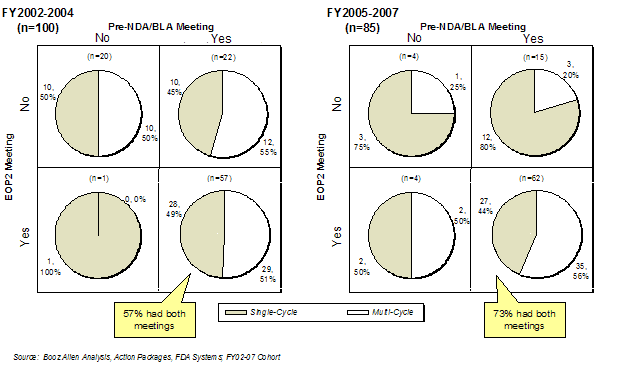
Nearly three-quarters (73%) of product applications had both a Pre-NDA/BLA and EOP2 meeting in the Prospective Analysis Cohort, which was a significant increase from the Retrospective Analysis Cohort (57%). Interestingly, products that had both meetings had a lower first-cycle approval rate (44%) than products that had only one or neither meeting (Exhibit 19). Further analysis regarding sponsor experience and impact on first-cycle approval is discussed in Sponsor Characteristics.
Exhibit 19. Incidence of Pre-NDA/BLA and EOP2 Meetings
FDA evaluates the application within the first 60 days of its receipt to determine if it is sufficiently complete to conduct a full review. If FDA determines that the application can be filed (i.e., the application is sufficiently complete), the application review continues. Under PDUFA III, FDA agreed to communicate to applicants any significant review deficiencies identified during the filing review by day 74 of the review cycle. Sending the Filing Review Notification was also specified as an activity in the Filing Review phase of the GRMPs. FDA considers the Filing Review Notification, commonly referred to as the 74-Day Letter, a preliminary review. FDA does not consider this review to be comprehensive nor indicative of deficiencies that may be identified later in the review cycle [12]. Similarly, FDA may not necessarily communicate deficiencies previously identified prior to the Filing Review (e.g., issues previously communicated during EOP2 or Pre-NDA/BLA meetings).
The 74-Day Letter was implemented to comply with PDUFA III goals in early 2003[13] (Exhibit 20). The 74-Day Letter was evaluated for effectiveness as a tool to provide earlier communication of issues to sponsors.
Exhibit 20. Filing Review Issues Identified by Year
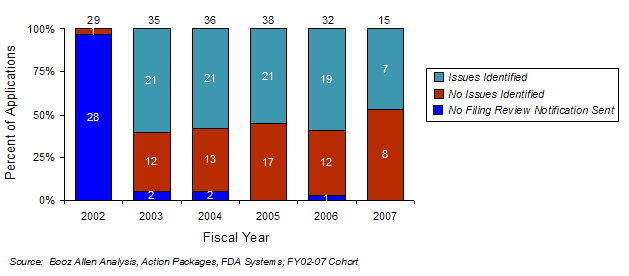
Since implementation of the 74-Day Letter, the number of letters with no issues identified has remained fairly constant. This suggests that upon adoption, FDA immediately began using this new tool for its intended goal of early sponsor communication, instead of sending a letter (e.g., with no issues identified) primarily to comply with the PDUFA III goals. Significant deficiencies were identified in 59% (89 of 152) of the applications for which a 74-Day Letter was sent to the sponsor (Exhibit 21). Although the filing review is a preliminary and non-comprehensive review, it appears to provide early identification of applications that are at risk for not being approved in the first cycle.
Exhibit 21. Impact of Issue Identification in Filing Review Notification for FY2002-FY2007 Cohort
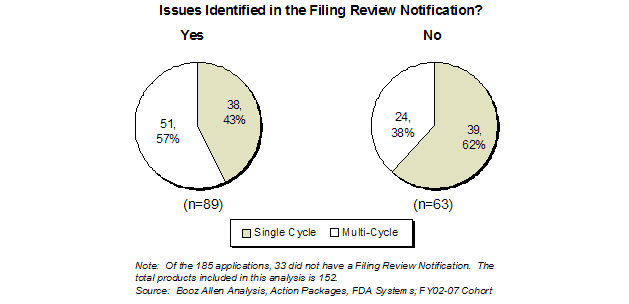
Of the 89 applications that had deficiencies identified in the 74-Day Letter, only 43% were approved in the first cycle, whereas 62% of applications with no significant deficiencies identified during the review were approved in the first cycle. Of the 24 multi-cycle applications that did not have any issues documented in the 74-Day Letter, 10 had issues identified in EOP2 or Pre-NDA/BLA meetings, and 14 had issues identified during the review.
The 74-Day Letter was an effective tool for early communication and issue resolution for those applications approved in a single cycle as well as those applications that were not approved in the first cycle. In the Prospective Analysis Cohort, 62% (29 of 47) of applications had all their potential review issues identified in the 74-Day Letter resolved by the action date (Exhibit 22). Of those that resolved all potential review issues identified in the 74-Day Letter, 62% were approved in the first cycle, indicating that the filing review successfully identified and communicated most of the major review issues to the sponsor in time to resolve them. As expected, a much smaller percentage (22%) of those applications with issues remaining unresolved from the 74-Day Letter at the action date were approved in the first cycle. For these approved applications, the unresolved review issues were not significant enough to negate the overall finding that the product was safe and effective, and were addressed as postmarketing study commitments.
Exhibit 22. Applications with All Filing Review Issues Resolved by Action Date for FY2005-FY2007 Cohort
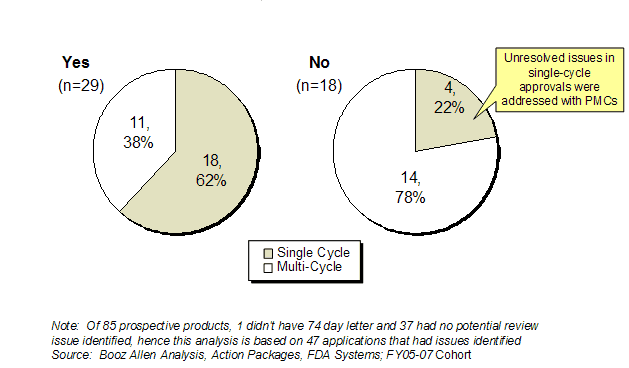
Many of the issues identified in the 74-Day Letter can impact approvability. As the data on 74-Day Letters indicate, the resolution of potential approvability issues during the review process is related to higher levels of first-cycle approval. The case study below, which evaluates the potential for an increase in the first-cycle approval rate, further illustrates the importance of addressing these issues during the review cycle when feasible. (Exhibit 23)
Exhibit 23. Approvability Impact of 74-Day Letter Issues
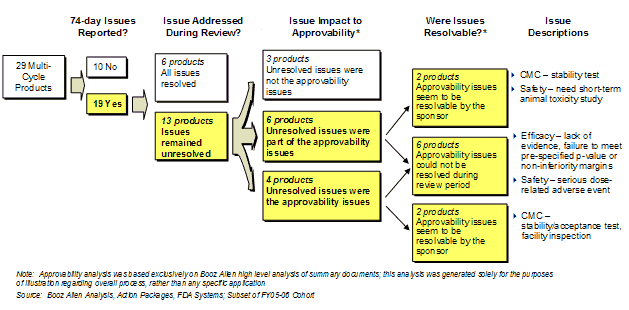
A subset of 29 multi-cycle applications from FY2005 and FY2006 was analyzed to illustrate the impact of 74-Day Letter issues that were not resolved. In this case study group, 19 of the applications had issues reported on the 74-Day Letter, and 13 of the applications did not resolve the issue by the Action Date. In 10 of these cases, the issue directly impacted the approvability of the application, which included CMC, efficacy, and safety issues. This case study is not necessarily quantitatively representative of the Overall Study Cohort, but rather illustrates that the 74-Day Letter often identifies important issues that impact approvability, and that many of these issues could be addressed during the timeframe of the first-cycle review.
The FDA and sponsors frequently engage in informal communications (e.g., email, telephone, and fax) in addition to face-to-face meetings during review of a product application. Sponsors generally contact FDA to determine the status of their product’s review or to respond to an information request while FDA contacts sponsors to request critical information and provide sponsors with opportunities to justify their findings and conclusions.
Exhibit 24. FDA-Sponsor Communications for FY2005-FY2007 Cohort
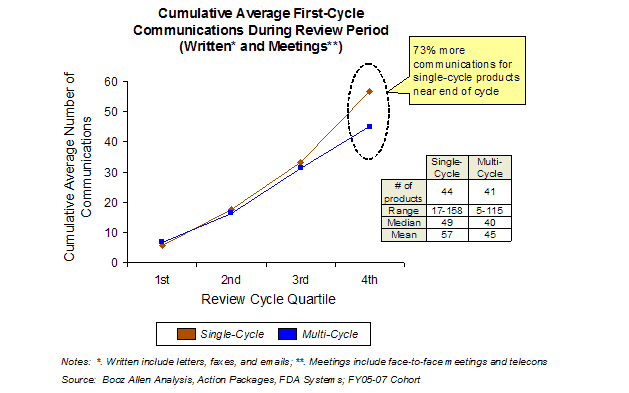
Analysis of Action Packages and FDA systems revealed that the frequency of FDA-sponsor communications was similar for single-cycle and multi-cycle reviews for the first three-quarters of the cycle (Exhibit 24). However, in the final quarter of the review cycle, there was a significant increase in communications for single-cycle approvals compared to multiple-cycle applications. Approximately 70% of these additional communications were related to labeling and PMC issues, suggesting that likelihood of approval drives the additional communications, rather than the reverse. This pattern was similar for both Standard and Priority review applications.
Further analysis of the FDA-sponsor communications showed that the overwhelming majority of communications used teleconference or written formats (Exhibit 25). According to an FDA guidance, FDA minimizes the use of resource-intensive face-to-face meetings during the review, which are instead reserved for products with specific issues as outlined in the relevant guidance.[14]
Exhibit 25. Communication Type for FY2005-FY2007 Cohort
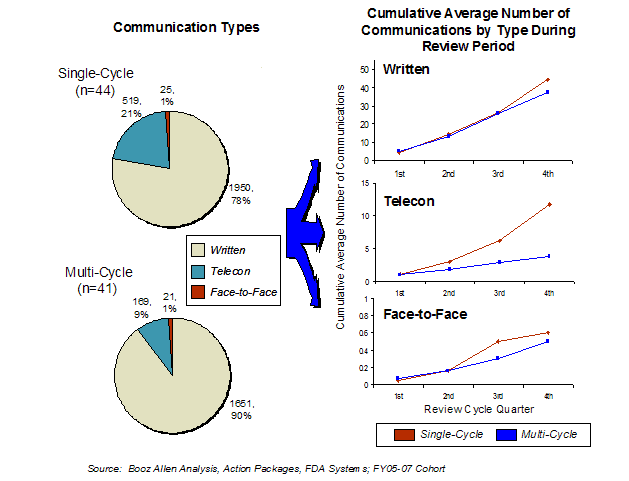
Most product applications (61%) did not have any face-to-face meetings during the review cycle. Booz Allen speculates the review teams focus on quickly resolving the outstanding issues for potential single-cycle products toward the end of the review by using teleconferences.
When sponsors respond to FDA's information requests, they submit written amendments to the NDA or BLA application. Sponsors also submit amendments to provide the FDA with additional safety (120-day safety update) and efficacy information that may have been agreed upon in earlier discussions. FDA generally asks for timely submission of these amendments to avoid disruption of the application review.
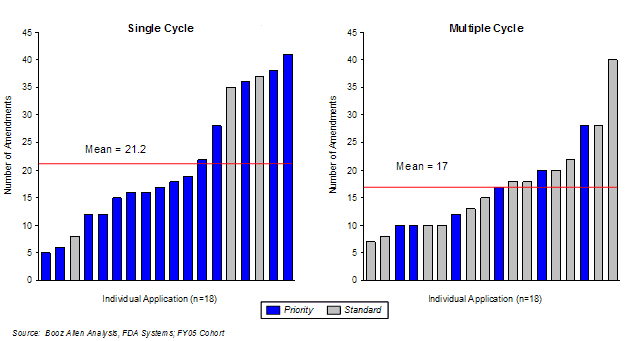
Booz Allen performed a case study on those sponsors that submitted amendments in FY 2005. Sponsors submitted more amendments, on average, for first-cycle approval products (21.2 per application) than for those that were not approved in the first cycle (17 per application), for all product applications submitted in FY2005 (Exhibit 26). There was no discernible difference between the number of amendment submissions for Priority or Standard review applications. The difference in the number of amendments submitted was almost entirely due to submissions in the fourth quarter of the review cycle. Booz Allen speculates that as with FDA-Sponsor communications and meetings, more amendments are also submitted when a product is nearing the approval Action Date.
Exhibit 26. Amendments Submitted for Applications for FY2005 Products
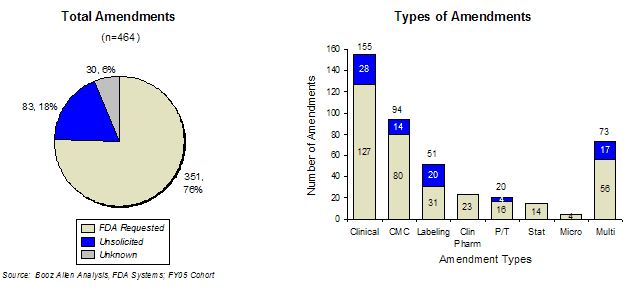
While amendments submitted by sponsors are typically in response to specific requests from FDA, sponsors may also submit amendments without prompting from FDA in order to supplement their application. In the FY2005 cohort of applications analyzed, 18% of all documented amendments were submitted without an FDA request for information. The largest proportion of amendments were related to clinical issues, followed by those regarding CMC and labeling issues.
Exhibit 27. Types of Amendments Submitted for FY2005 Applications
Advisory Committee meetings provide FDA with an additional opportunity to discuss deficiencies of an application by soliciting independent, external advice from experts knowledgeable in specific areas related to drug and biologic products. Although the committee members provide advice to FDA and may recommend approval or disapproval of the application, the FDA is not bound to follow the recommendations of the Advisory Committee (Exhibit 28).
Exhibit 28. Advisory Committee Meetings and Review Designation
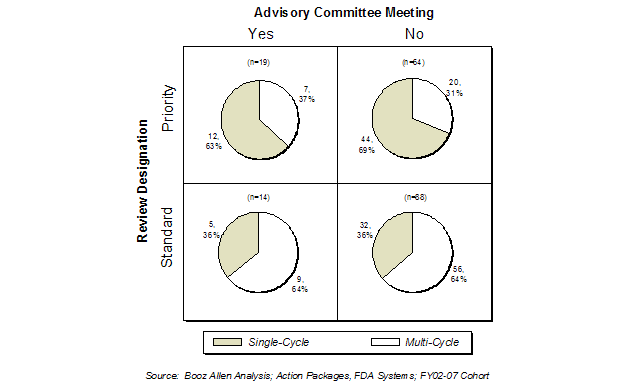
In the FY2002-FY2007 cohort, only 18% of the product applications had Advisory Committee meetings. The presence of an Advisory Committee meeting did not seem to impact the likelihood of an application being approved in the first cycle, regardless of whether it was a Priority or Standard review application.
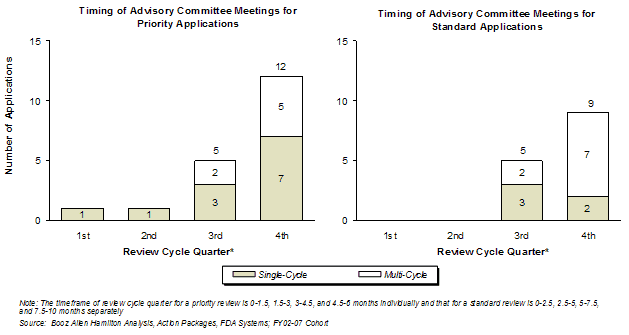
Most Advisory Committee meetings took place in the latter half of the review cycle (Exhibit 29). The timing of the Advisory Committee meeting did not impact the first-cycle approval rate for Priority review applications. With the limited data, a slightly greater proportion of Standard review applications with an Advisory Committee meeting in the fourth quarter were not approved in the first cycle than those with the meeting in the third quarter of the review cycle.
Exhibit 29. Advisory Committee Meeting Timing and Priority/Standard Designation
Applications with major deficiencies [15] identified and documented either pre-submission or during the review were less likely to be approved in a single-cycle than those applications that did not have a major deficiency identified during the same timeframe (Exhibit 30).[16]
Exhibit 30. Impact of Major Issues Identified on Approval Rate in FY2005-FY2007 Cohort
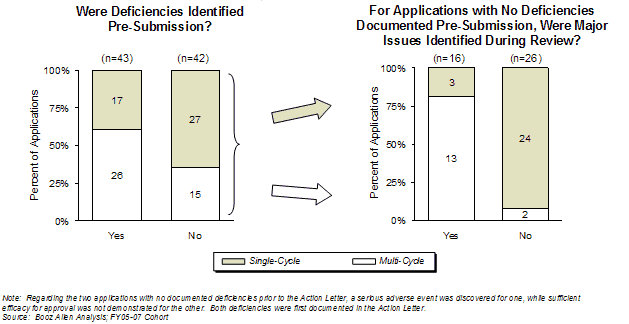
However, applications were more likely to be approved in the first-cycle if a major deficiency was identified pre-submission (40%) than if major deficiencies were only found during the review (19%). Applications for which no major deficiency was identified either pre-submission or during the review had a higher first-cycle approval rate (92%). The two multi-cycle products that did not have deficiencies identified pre-submission or during review had either marginal therapeutic benefit or a greater risk/benefit profile at the end of the review cycle.
The majority of multi-cycle applications have major deficiencies in only one or two categories (Exhibit 31), however, these were in the critical areas of safety, efficacy or chemistry, manufacturing and controls.
Exhibit 31. Major Deficiencies Cited in First Action Letter of Multi-Cycle Applications by Category for FY2002-FY2007 Cohort
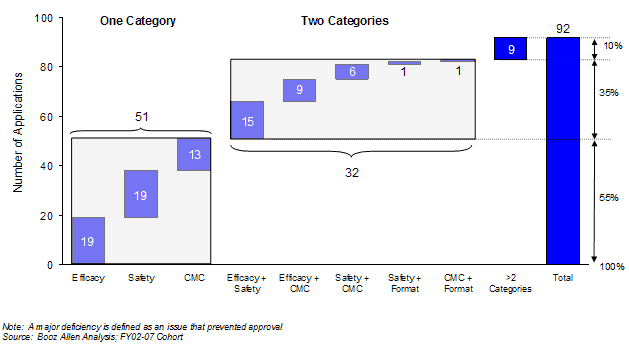
Of the 92 applications requiring multiple cycles, 51 (55%) were cited for a single significant deficiency in the safety, efficacy or CMC categories. Thirty-two applications failed due to deficiencies in a combination of two of these categories, and two for a combination of application format and either CMC or safety. The nine remaining multiple cycle applications failed with significant deficiencies in more than two categories.
These deficiencies were further categorized into the areas of design (e.g., trial or manufacturing process design), execution (e.g., unacceptable clinical execution), or failure to meet study objectives (e.g., clinical endpoints) (Exhibit 32).
Exhibit 32. Major Deficiencies Cited in First-Cycle Action Letter of Multi-Cycle Applications by Area for FY2002-FY2007 Cohort
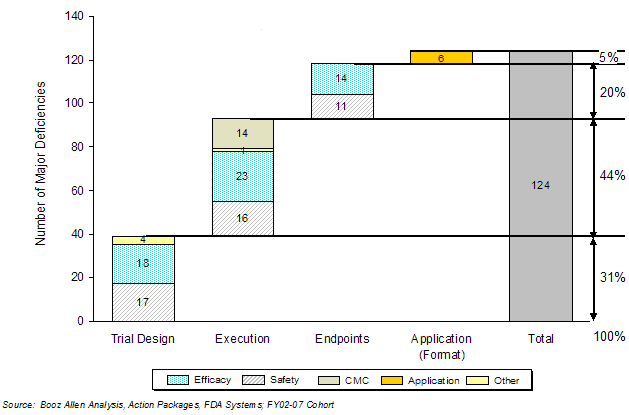
Of the 124 major deficiencies cited in 92 first-cycle action letters, 39 were related to trial design, 54 to execution and 25 to endpoints. The remaining six application format deficiencies were related to inconsistent documentation or record keeping, inability to locate information, or failure to translate from foreign languages into English. Exhibit 33 provides examples of the types of application issues that were identified by category type.
Exhibit 33. Examples of Issues Observed by Category
| Issue Category | Design | Execution |
|---|---|---|
| Efficacy |
|
|
| Safety |
|
|
| CMC |
|
|
| Format (primarily in missing data) |
|
Postmarketing study commitments (PMCs), also referred to as Phase 4 commitments, are studies that are conducted by a sponsor after FDA has approved a product for marketing. FDA requires PMCs for products in certain situations, such as those approved under the accelerated approval provision, based on animal efficacy data, or not sufficiently labeled for pediatric use [17].
Exhibit 34. Products with Postmarketing Study Commitments in FY2002-FY2007 Cohort
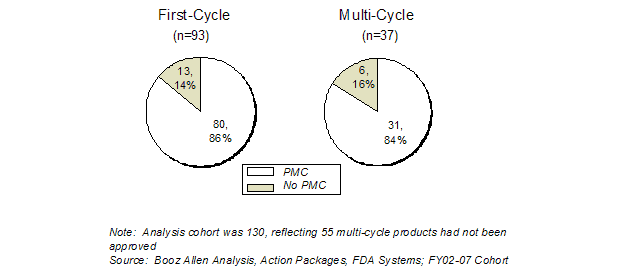
Agreed-upon PMCs are intended to further characterize the safety, efficacy, or optimal use of a product, or to ensure consistency and reliability of product quality. Agreed-Upon PMCs can be used to resolve important issues that do not override the determination that the product is effective and safe for marketing. For the FY2002-FY2007 cohort products, nearly the same proportion of single-cycle (86%) and multi-cycle (84%) applications were approved with PMCs (Exhibit 34). While the percentage of applications with PMCs was similar between single and multi-cycle, there was a significant difference in the number of PMCs assigned.
Exhibit 35: Distribution of PMCs for Single- and Multi-Cycle Approvals
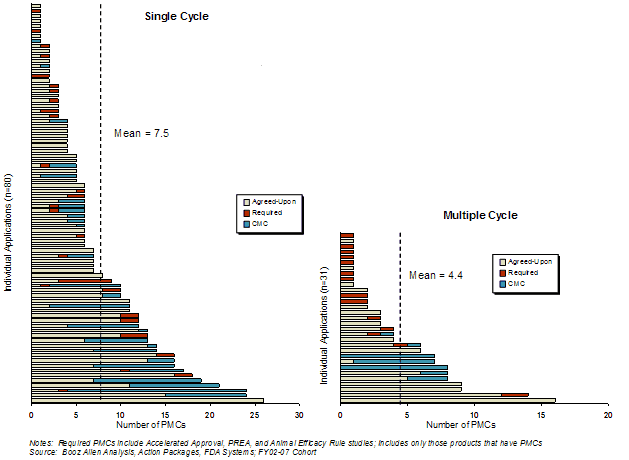
The average number of PMCs associated with applications that had PMCs was significantly different for single (7.5) and multiple cycle (4.4) review products (Exhibit 35). This difference was primarily due to a greater number of required and CMC PMCs associated with single-cycle applications. There were a similar number of agreed-upon PMCs for single (5.4) and multiple (4.6) cycle review applications. [18].
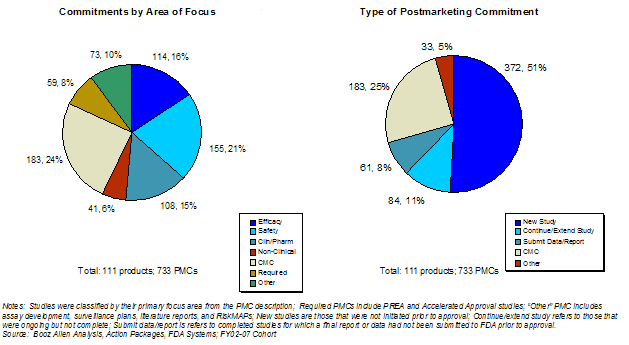
Exhibit 36 shows that the greatest proportion of PMCs are related to CMC issues (24%), followed by clinical safety (21%), clinical efficacy (16%) and clinical pharmacology (15%). A new clinical study request constituted 51% of PMCs. A detailed analysis of agreed-upon PMCs and their associated development and tracking processes was conducted in a separate study commissioned by FDA and completed in 2007.
Exhibit 36. Focus Area of Postmarketing Study Commitments for FY2002-FY2007 Cohort
Booz Allen investigated sponsor characteristics as factors for influence on first-cycle review rates. Sponsors were characterized by their experience (e.g., prior approvals with FDA in general and in same therapeutic class), the size of the company based on market capitalization, the type of company (pharmaceutical or biotechnology only), and origin based on location of headquarters. Based on observations during the study, many of these factors tended to have a direct or indirect impact on the sponsor’s ability to respond to FDA information requests, either through resource availability or knowledge of FDA policies and procedures. In turn, the sponsor’s ability to respond to FDA’s information requests can impact approvability.
Sponsor experience with the FDA approval processes appears to contribute to first-cycle approvals. Experienced sponsors had first-cycle approval rates of 55% compared to 38% for sponsors with no prior approved products (Exhibit 37).
Exhibit 37. Single-Cycle Approval Rate by Sponsor Experience
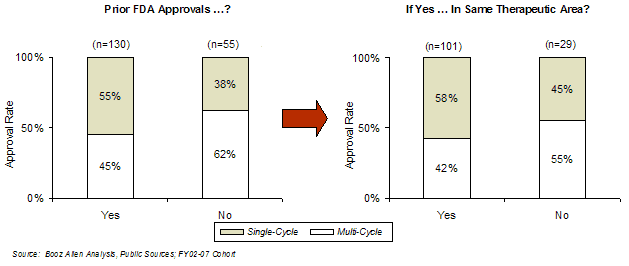
For those sponsors that had prior experience, the first-cycle approval rate was greater if that experience was in the same therapeutic area (58%) than if it was in a different area (45%). This result is consistent with feedback from FDA focus groups, which suggested that unfamiliarity with FDA regulations and the drug/biologic application process is a key problem for inexperienced sponsors and results in poor quality submissions.
Of sponsors with no prior FDA approval experience, 52% opted not to hold an EOP2 meeting compared to 28% of experienced sponsors. For the Pre-NDA/BLA meeting, 20% of inexperienced sponsors did not have the meeting, compared to 14% of experienced sponsors. Despite lower overall approval rates, inexperienced sponsors did not take advantage of opportunities to meet with FDA, where they might have been able to learn and resolve key application issues prior to submission (Exhibit 38).
Exhibit 38. Sponsor Experience and Impact of Pre-Submission Meetings
Booz Allen also examined the amount of communication based on sponsor experience. Sponsors differed in the amount of documented communication they had with FDA throughout the review cycle, depending on their level of experience.
Exhibit 39. Cumulative Average First Cycle Communications During Review Period by Sponsor Experience
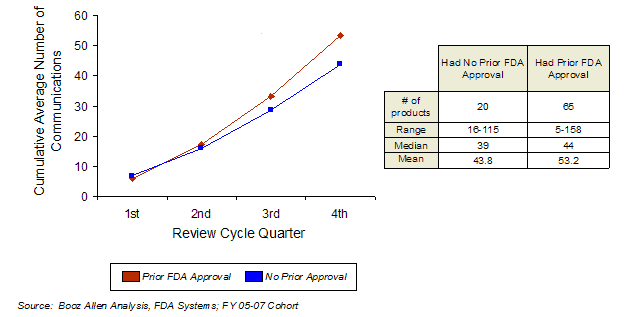
Sponsors that had prior FDA approvals averaged more communications during the review cycle, with the increase occurring primarily during the last half of the review. These findings are consistent with previous findings that sponsors with applications moving towards approval have more communication with FDA at the end of review to discuss labeling and postmarketing commitments.
Large sponsors have usually experienced a successful FDA application submission in the past, so it is not surprising that large sponsors had higher first-cycle approval rates. Applications submitted by large sponsors (i.e., market capitalization greater than $5 billion) were more likely to gain first-cycle approval for both traditional pharmaceutical and biotechnology companies than were medium- or small-sized (i.e., market capitalization under $5 billion) pharmaceutical or biotechnology companies (Exhibit 40). Booz Allen speculates that the larger companies have more resources and processes dedicated to supporting the FDA review process than smaller companies.
Exhibit 40. Approval Rate vs. Sponsor Type and Origin
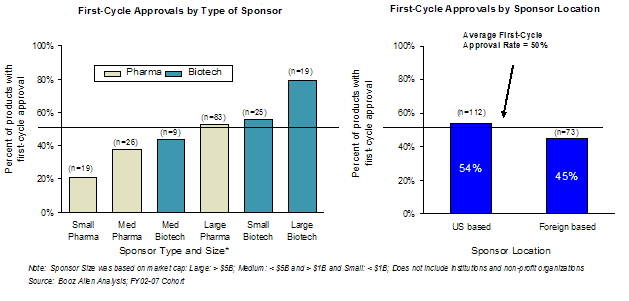
Overall, biotechnology companies in the study had higher rates of first-cycle approval for their products than did pharmaceutical companies. This difference appears to be largely due to the fact that biotechnology companies in the Overall Study Cohort had a greater proportion of Priority review applications (55%) among their total applications than did pharmaceutical companies (40%). The first-cycle approval rates were also slightly higher for US-based sponsors (54%) than foreign sponsors (45%).
While there were differences in review designations between pharmaceutical and biotechnology companies that impacted the first-cycle review rates, both large and small companies had approximately equal proportions of Priority and Standard review applications in the full cohort (Exhibit 41).
Exhibit 41. First Cycle Approval Rate by Review Designation and Company Size
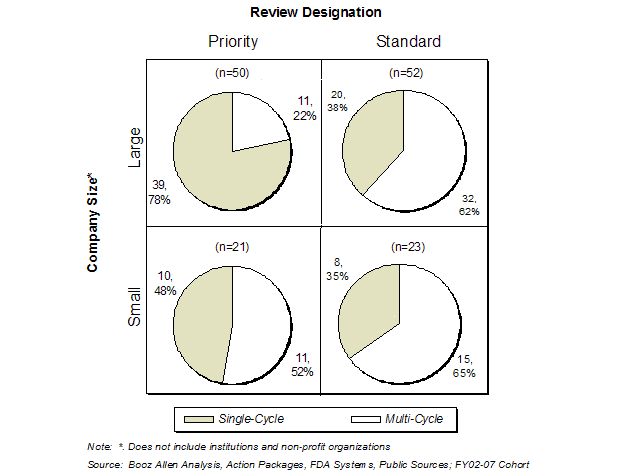
The approval rate for Standard review applications was similar regardless of company size (38% for large vs. 35% for small). However, the difference in approval rate on Priority applications was much more significant (78% for large vs. 48% for small), and accounts for the greater first-cycle success for large companies. Booz Allen speculates that this substantial difference may indicate that smaller companies have more deficiencies in their applications and have difficulty managing the compressed, six-month review timeframe of the Priority review, while larger companies with more resources are better able to respond to FDA concerns and issues in a timely fashion.
Further analysis of a company’s size and application deficiencies revealed a noticeable trend in the number of categories of deficiencies identified and documented in the first action letter of multi-cycle products (Exhibit 42). Small company applications were divided nearly evenly between those with deficiencies in one, two or more than two categories. Medium companies had deficiencies in two categories in just over half of their multi-cycle application Action Letters and did not have any action letters citing more than two categories of deficiencies. Finally, nearly two-thirds of large company action letters of multi-cycle products had only one category of deficiency and there were no action letters with more than two categories. This illustrates that within the Prospective Analysis Cohort, even among applications that were not approved in the first cycle, larger companies had fewer areas with significant outstanding issues, suggesting that they either had better quality submissions or were more successful at resolving issues prior to the Action Date.
Exhibit 42. Deficiencies in Multi-Cycle Products by Sponsor Size in FY2005-2007
| Company Size | One Category | Two Categories | More Than Two Categories |
|---|---|---|---|
| Small (n=11) |
4 products
|
4 products
|
3 Products |
| Medium (n=8) |
3 Products
|
5 Products
|
None |
| Large (n=22) |
14 Products
|
8 Products
|
None |
When further examining the differences between small and large companies, there appear to be differences in the number of communications initiated by either FDA or sponsors. Larger companies average more communications with FDA regarding their applications during the first-cycle review than either small or medium companies (Exhibit 43), particularly in the first half of the review cycle.
Exhibit 43. Cumulative Average First-Cycle Communications During Review Period by Company Size
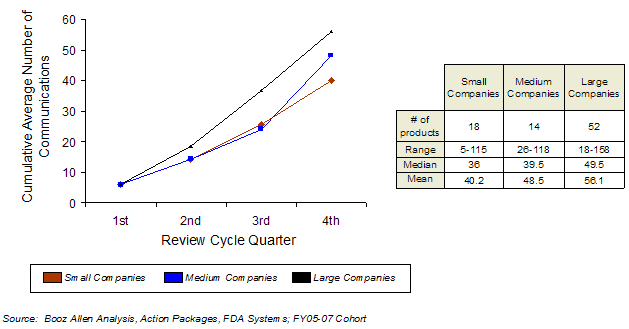
In addition to the findings already discussed concerning end-of-review communication (i.e., labeling and PMC discussion), Booz Allen speculates that this greater amount of early communication may help larger companies respond to FDA concerns and resolve issues more rapidly. Booz Allen hypothesizes that an increase in resources available to communicate with FDA may explain the higher rate of first-cycle approval observed for large companies than in small or medium companies. In particular, Booz Allen speculates this may help facilitate better outcomes in compressed Priority review schedules, which is consistent with the higher first-cycle approval for larger companies with Priority applications.
Booz Allen analyzed first-cycle approval rates by Offices (along with other factors) to identify best practices in application review. Booz Allen also studied the number of applications by office, staffing changes and manufacturing inspections to determine their effects on first-cycle approval rates. These factors were analyzed independently without other potential factors that might affect first-cycle approval (e.g., application quality)
Product approval was analyzed by review office to identify trends. The total number of applications was analyzed as well as the total number of Priority and Standard review designated applications. The first-cycle average approval rate by review office was analyzed and compared to the average overall cohort first-cycle approval rate. Additional analyses of average first-cycle approval rates by office for Priority and Standard designations were conducted (Exhibit 44).
Exhibit 44. Cohort Applications by Office and Review Designation
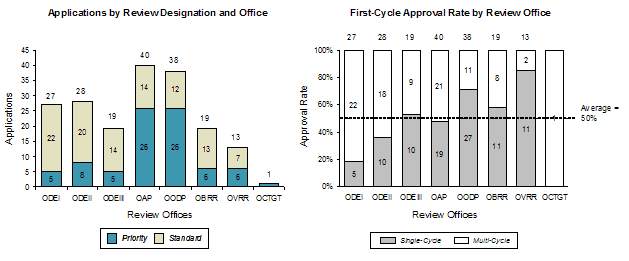
The analysis of application distribution by review offices revealed a range from 1 application to 40 applications per office over the FY2002-FY2007 cohort. The Office of Antimicrobial Products (OAP) and the Office of Oncology Drug Products (OODP) had the largest number of applications (40 and 38, respectively). OAP and OODP also had the largest overall number of Priority-designated applications, with each having 26 applications.
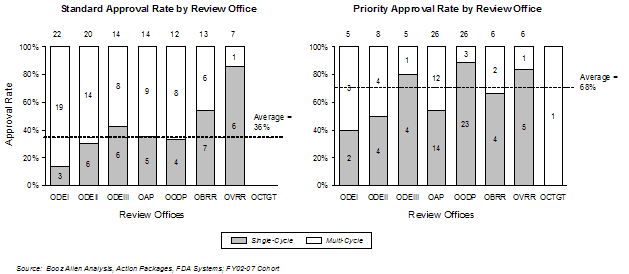
The overall first-cycle approval rate by review office analysis showed that three of the four offices with more than 20 products in the FY2002-FY2007 timeframe had average approval rates lower than the overall cohort average of 50% (Office of Drug Evaluation(ODE) I, ODE II, OAP were lower, OODP was higher than average). The highest overall first-cycle approval levels were found in OODP and Office of Vaccines Research and Review (OVRR), while the lowest were found in ODE I, ODE II, and Office of Cellular, Tissue, and Gene Therapies (OCTGT).
The average overall first-cycle approval rate for Standard applications was 36% for FY2002-FY2007; for Priority approval products it was 68%. In general, the first-cycle approval rate in an office was correlated with the proportion of Priority review applications in the office. The only notable exceptions were ODE III and OVRR, which had higher approval rates than would be expected based on the number of Priority applications, and OAP, which had a lower approval rate than would be indicated by the proportion of Priority applications.
The number and timing of application submissions were evaluated as indicators of workload since FDA does not utilize 100% time reporting to track the duration of time spent reviewing a particular application and supporting activities.
Exhibit 45. Submission Timing and First Cycle Approval Rates
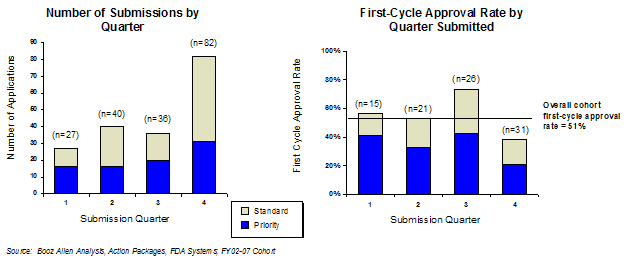
Forty-four percent of all applications were submitted in the fourth quarter of the fiscal year throughout the cohort period, more than twice as many as the next highest frequency quarter (Exhibit 45). The first-cycle approval rate was also the lowest for applications submitted in the fourth quarter, and the only quarter that was below the study average (38% in the fourth quarter vs. 51% overall). Examination of other factors (e.g., review designation, sponsor size, sponsor experience) did not provide any insight regarding the significant differences with these applications in comparison to applications submitted in other quarters.
Feedback in focus groups with FDA review staff suggested that staffing changes in the management or review team could cause inefficiencies in the review process. This hypothesis was tested in the FY2002-FY2005 cohort, by analyzing first-cycle approval rates as a function of staff changes of Office leadership, Division leadership, Medical team lead, and RPM. [19]
Exhibit 46. Staffing Changes Between Pre-Submission and Review
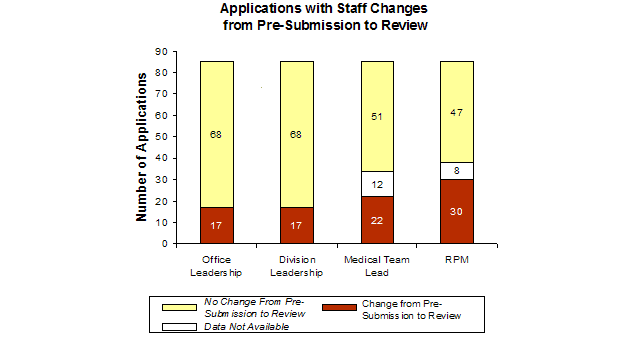
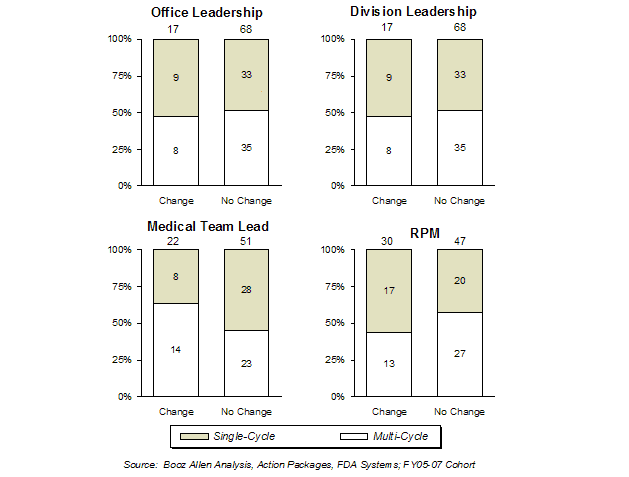
Across the cohort, the same staff member was generally involved with the application from pre-submission through the review. When staff turnover occurred the RPM was the most common position change (38% RPM change for applications where information was available). Changes in staff between pre-submission and review did not seem to impact the first-cycle approval rate, except for the Medical team lead turnover (56% with no change, 38% with change). Based on limited data, staff turnover did not impact first-cycle approval rates.
Current Good Manufacturing Practice (cGMP) compliance is integral to the review process and action. For an efficient review, FDA reviewers stressed the importance of effective internal communication with divisions overseeing manufacturing compliance. Interviewees stated that delays in cGMP inspections can slow the review process and/or result in multi-cycle reviews. Manufacturing deficiencies uncovered late in the review cycle may not allow sponsors sufficient time to correct issues before the Action Date. This concern was particularly pronounced for applications requiring inspections at foreign locations which, due to increased administrative requirements as well as field inspector resource constraints, generally have longer lead times.
There was no significant difference observed in the first-cycle approval rate between applications requiring foreign manufacturing site inspection and those with only domestic manufacturing inspections, as shown in Exhibit 47.
Exhibit 47. Impact of Foreign or Domestic Manufacturing Site
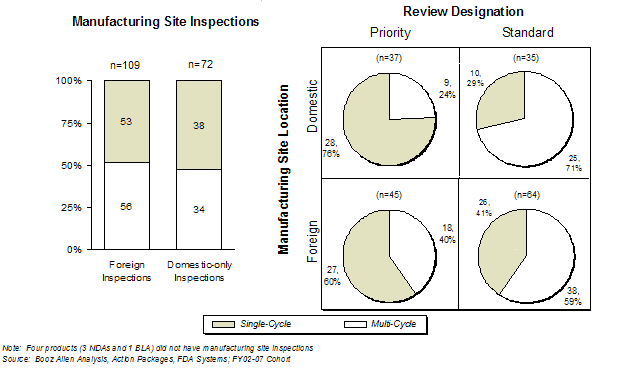
These data suggest that despite the potential for delay due to logistical challenges of foreign inspection, FDA manages this process efficiently, without significant delay that impacts first-cycle approval. However, the long lead times for the planning and execution of site inspections (up to four months, with additional vulnerabilities for foreign inspections) can place single cycle approvals at risk for applications with Priority designation, which have compressed review times. Among Priority review products, the first-cycle approval rate for applications requiring a foreign inspection was lower (60%) than for those with only domestic site inspections (76%), suggesting that the compressed review time may impact first-cycle approval when foreign manufacturing inspections are needed. Indeed, either an inability to conduct or failure of a manufacturing site inspection contributed to the lack of first-cycle approval for one-third of the multi-cycle Priority review applications that required a foreign manufacturing site inspection. [20]
Booz Allen developed recommendations based on the analysis and findings for the Overall Study Cohort combined with insights gained during our interviews and focus groups with FDA and Sponsors. In general, our recommendations can be classified as either those that are designed to improve the quality of the submission or those designed to improve the quality of the review process (Exhibit 48).
Exhibit 48. Summary Overview of Recommendations
| Recommendation | |
|---|---|
| Pre-Submission |
1. Meeting Focus: FDA should continue its recent shift in emphasis on Pre-NDA/BLA meetings to include content in addition to format of product application. 2. Quality Measures: FDA should develop application quality measures to encourage future submission quality by offering sponsors an application checklist. |
| During Review and Post-Action |
3. Communications Protocol: Sponsors and FDA should develop and obtain agreement on an application review communication protocol upon submission (e.g., communication format). 4. GRMPs Implementation: FDA should continue to implement GRMPs activities and timeframes for application review. 5. Quality System: FDA should supplement the GRMPs guidance with a quality system approach to continuously monitor and refine the process. |
| Overall |
6. Early Issue Communication: FDA should continue early communication of issues through pre-submission meetings (e.g., EOP2, Pre-NDA/BLA) and during review (e.g., information requests, 74-Day letter). 7. Tracking and Resolution of Issues: Sponsors should continue to take advantage of opportunities for FDA feedback on potential and actual application issues such as EOP2, Pre-NDA/BLA, and 74-Day Letter Review Issues; sponsors should actively track and resolve these issues prior to application submission. 8. FDA Outreach: FDA should proactively initiate workshops with sponsors to outline communication methods, guidances, and FDA tools available throughout product development. 9. FDA Website: FDA should display a prominent tab on the FDA home page that organizes critical information and guidance for sponsors of new drugs and biologics. 10. Documentation Manual of Policies and Procedures (MaPP): FDA should develop a MaPP outlining best practices for documenting internal meetings and sponsor interactions. 11. Documentation of Sponsor Interactions FDA should consistently document sponsor interactions that are likely to impact product approvability (e.g., information requests, pre-submission conversations). 12. Knowledge Sharing Tool FDA should develop/use an internal FDA knowledge sharing repository to assist reviewers with approaches to resolve review issues. |
Note: Some Offices already have some of these concepts in place.
FDA provides many opportunities for sponsors to interact with and obtain feedback from FDA prior to and during the application review process. These interactions represent valuable opportunities for the sponsor to obtain feedback regarding potential issues. Sponsors should continue to participate in these meetings. Booz Allen observed higher first-cycle approval rates in applications submitted within six months of having the Pre-NDA/BLA meeting for the FY05 – 07 cohort than in the FY02 – 04 cohort. FDA should continue its shift in emphasis of discussing data, in addition to application format issues.
Developing approaches for improving application quality will be the next challenge for FDA and sponsors after structured review processes have been successfully adopted. Activities such as developing quality measures based on post-action application assessment and development of a quality checklist for sponsors to self-evaluate prior to submission may improve application quality.
Based on the findings, there is a significant difference in the frequency of communication associated with first-cycle vs. multi-cycle applications. However, interviews with FDA staff indicate many sponsors engage FDA in non-productive communication via phone and email to determine the status of application. To improve the efficiency and the effectiveness of the review process, Booz Allen recommends that FDA and sponsors agree on an application review communication protocol during filing determination. Such a communication protocol would set expectations for sponsors about appropriate methods (e.g., secure email) and timeframes for FDA communication.
Although GRMPs have not yet been fully implemented, preliminary findings indicate that application reviews with high levels of compliance with assessed GRMPs activities had high first-cycle approval rates. As such, Booz Allen recommends that FDA continue forward with GRMPs implementation, ensuring adoption of important milestones and timeframes. Based on an initial 2006 assessment of GRMPs adoption and ongoing implementation, the impact of GRMPs should be assessed after rollout is completed. Additionally, GRMPs need a mechanism for process improvement and feedback through quantitative metrics and staff perception, which could be provided through a quality system approach. The objective of the quality system is to develop the mechanisms needed to sustain and improve the GRMPs. The quality system would include methods to evaluate the effectiveness of the GRMPs and provide processes and procedures to monitor, maintain, and refine the GRMPs.
Pre-submission meetings identified significant issues that ultimately impacted application approvability. FDA should continue to encourage sponsors to request these meetings and resolve the issues prior to application submission. Booz Allen findings indicate that the 74-Day Letter is an effective tool for FDA to communicate application issues. As indicated previously, the presence and severity of application issues was a major factor in multi-cycle review. The proactive identification of application issues through the 74-Day Letter allowed applicants in many cases to address application issues prior to action. The FDA should continue to use the 74-Day Letter as an effective tool to communicate application issues.
Sponsors should maximize the efficiency and the effectiveness of the review process by tracking and resolving the issues identified during pre-submission meetings (e.g., EOP2, Pre-NDA/BLA) prior to application submission and actively resolving those issues identified during the review phase to the extent practicable. Sponsors should continue to acknowledge and address the issues presented in the 74-Day Letter. Further, sponsors need to recognize that the 74-Day Letter does not represent a comprehensive listing of all application issues identified previously (e.g., EOP2 or Pre-NDA/BLA) or those that may be identified subsequently during the review process.
Inexperienced sponsors were less likely to hold key pre-submission meetings and had fewer FDA interactions throughout the review. These sponsors would likely benefit from FDA-initiated efforts to clarify review processes and delineate tools and guidances available from FDA during product and application development. One way to obtain this information is to engage in early and open dialog employing FDA-preferred methods (e.g. appropriate forms and correct submission procedures), and develop processes to rapidly respond to FDA requests. The FDA can facilitate these activities with workshops and updated and streamlined guidance portfolios, as well as improving the utility of the website, which includes sections targeted to these sponsors. FDA could also update their website to better organize tools and guidances needed by sponsors during product development. Implementing and maintaining these recommendations may require additional FDA resources, but the cost of these additional resources could be offset, in the long-term, by reducing the incidence of multi-cycle reviews.
Booz Allen recommends that FDA develop a structured approach in the form of a MaPP for identifying and documenting FDA-sponsor interactions. FDA should consider using Customer Relationship Management (CRM) software to document interactions. Based on the importance of issue resolution in achieving first-cycle approval, FDA should develop a knowledge repository or sharing tool to identify, collect, and share successful experiences where issues were resolved during review. More disciplined internal documentation practices will help maintain the FDA’s knowledge of issues communicated and information requested regardless of staff transition or long product development timeframes (e.g., multi-cycle review). Developing a knowledge sharing capacity (e.g., cross application search capability to quickly locate information, location for knowledge exchange forums) among review staff will assist reviewers with identifying tested approaches and effective communication methods for issues across therapeutic areas. Many commercial off-the-shelf knowledge management products (e.g., Documentum, Vignette, and FileNet) offer a set of features (e.g., content authoring, content storage, publication management) that could meet FDA’s business needs.
GRMPs implementation began with the publication of guidance in April 2005 and is still continuing. Given this timing, only applications reviewed in the Prospective Analysis Cohort (FY2005-FY2007) were considered in measuring GRMPs compliance.
A baseline level for GRMPs implementation was collected through interviews with Division Directors and review staff held in January 2006. Changes in GRMPs adoption were assessed over time as each product in the Prospective Analysis Cohort was reviewed (FY2005-FY2007). Interview data was required because many of the GRMPs steps do not result in documentation that can be analyzed. The baseline assessment of FDA’s practices could be used to assess future levels of improvement in compliance (Exhibit 49). The baseline analysis showed that while the majority of GRMPs activities were adhered to, FDA Divisions were challenged to consistently meet timelines after the filing and review planning phase. FDA interviewees cited insufficient staffing to address the review workload as a primary factor in failure to meet GRMPs deadlines.
Exhibit 49. Baseline of GRMPs Practices Adopted by Divisions – January 2006 Snapshot
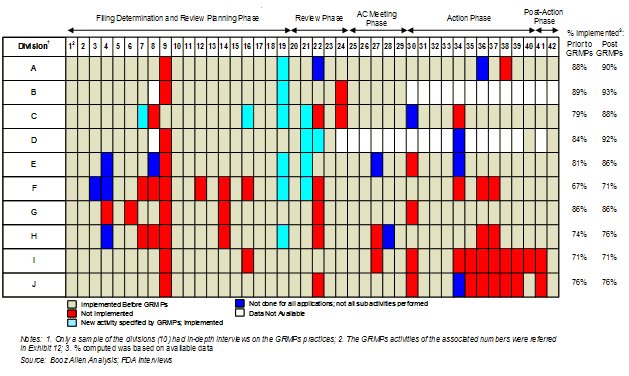
A product-by-product assessment of GRMPs adoption for the FY05 to FY07 cohort showed GRMPs adoption progressed at different rates across offices. For this cohort, 20% of the reviews (17 of 85) complied with 80% or more of the five assessed activities (Exhibit 50). [21] More than half (56%) of the reviews of products in the cohort complied with two or less of the measured activities [22].
Exhibit 50. GRMPs Compliance by Product
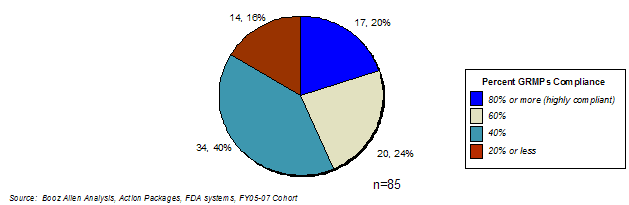
Most offices had started implementing GRMPs. However, the extent to which they had implemented them varies considerably. One office was highly compliant with 57% of its reviews. Of the remaining offices, high GRMPs compliance was not achieved with more than 25% of the application reviews (Exhibit 51).
Exhibit 51. CDER and CBER GRMPs Compliance by Office
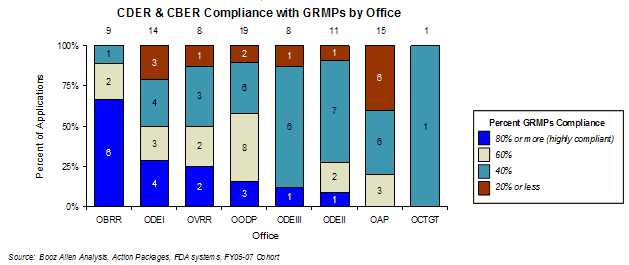
Improvement in compliance was analyzed for the period following GRMPs rollout (FY 2005- FY 2007). As GRMPs were developed from existing best practices in offices, some offices had high levels of initial compliance with activities. Compliance with GRMPs activities in the Review Phase increased substantially between FY05 and FY07. Mid-cycle meetings were conducted on time for just 8% of reviews in FY05, but for 63% of reviews in FY07 (Exhibit 52). Completion of the primary clinical review within the specified timeline also improved, from 16% in FY05 to 38% in FY07.
Exhibit 52. GRMPs Compliance in Review Phase
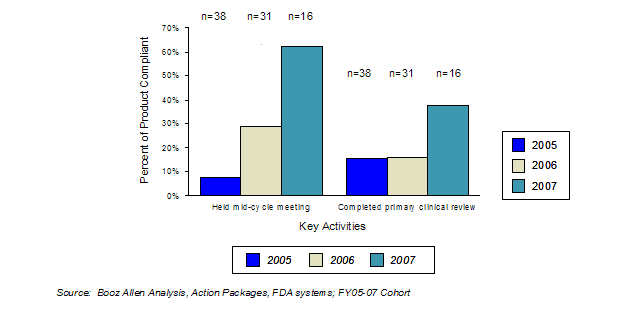
Yet, compliance with measured GRMPs activities in the filing and action phases had remained constant throughout the three-year period. Compliance with sending out the 74-Day letter was consistently over 90% throughout the assessed time period (Exhibit 53). The filing meeting was held within the specified timeline for approximately 60% of applications throughout the cohort period. Similarly, labeling discussions took place during the action phase in approximately 45% of application reviews in each of the three years.
Exhibit 53. GRMPs Compliance in Filing and Action Phases
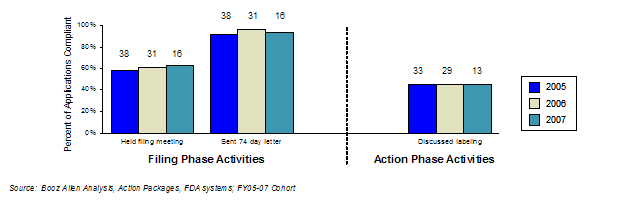
In March 2006, two industry focus groups were convened that represented 19 companies with recent product submissions. The objective of the industry focus group was to capture industry reaction to GRMPs and factors that contribute to or detract from first-cycle approval. The focus group participants spanned the pharmaceutical and biotechnology sectors with 47% representing large pharmaceutical companies, 21% representing large biotechnology, 16% representing small biotechnology and 16% representing small to medium Pharma. [23] The average self-reported regulatory experience of the participants was 14 years and in total spanned all CBER/CDER product divisions. Following the sponsor focus groups, two FDA focus groups were held to collect comparative perspectives from discipline reviewers, Medical team leads, chief Regulatory Project Managers, Regulatory Project Managers, branch chiefs and policy analysts.
Industry participants indicated they found varying levels of GRMPs compliance across Divisions. Few sponsors had made any internal process changes in response to the GRMPs issuance and even fewer had provided input to the draft GRMPs. The industry participants reported the GRMPs guidance and principles were valued. Further, the industry participants anticipated the GRMPs would enhance standardization of review communication practices across Divisions. Finally, industry participants felt it was too soon to assess the impact of GRMPs as the rollout is underway but some had used the guidance for internal education of management regarding FDA review practices.
Sponsors agreed that an effective communication plan with FDA is important and would help establish expectations, promote consistency across Divisions, and allow for needed review updates without adversely impacting FDA reviewers workloads (Exhibit 54).
Exhibit 54. Review Communication Practices Preferred by Industry and FDA Focus Group Participants

Industry participants believed review team consistency contributed to a good review and that Regulatory Project Manager experience was critical; they believed RPM changes (e.g., staff turnover) were detrimental to the review. When considering the filing and planning and review phases, sponsors saw value in the Application Orientation meeting [24] but believed the 74-Day letter was useful only if it contains major review issues. Industry participants believed their response time to identified issues took into consideration the severity of the issues and sometime was impacted by the need for FDA staff to clarify issues or needed actions (Exhibit 55).
Exhibit 55. Industry and FDA Focus Group Feedback on GRMPs Filing and Planning and Review Activities
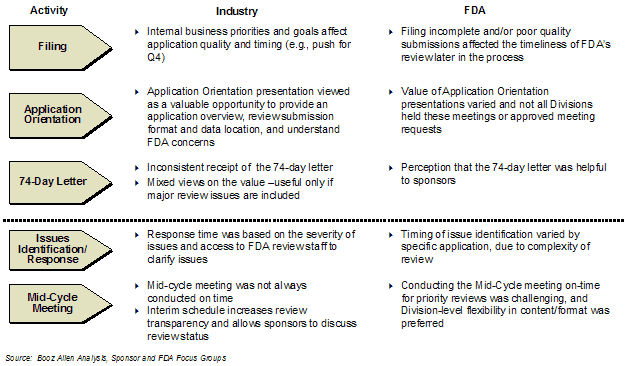
When considering the Advisory Committee, action and post-action phases, both FDA and sponsors agreed Advisory Committee meetings are resource intensive and especially challenging for the six-month, Priority reviews.
56. Industry and FDA Focus Group Feedback on GRMPs Advisory Committee, Action and Post-Action Phases

The metrics captured for hypotheses developed and tested in the First Cycle Approval Evaluation are detailed below by the following categories:
Each hypothesis is accompanied by the anticipated metrics for assessment, as well as the data sources that were used or evaluated for testing. Some hypotheses were tested, of these some had significant findings and others did not. Some hypotheses could not be tested, either because appropriate data did not exist or the quality and quantity of such data was insufficient. Each analysis is marked with a status that details the analysis outcome:
Product/Disease Characteristic Hypotheses
| Hypotheses | Metric(s) | Data Source | Status |
|---|---|---|---|
| 1. Products in the Fast-Track program have higher first-cycle approval rates |
|
|
AF |
| 2. Products with Priority review have higher first-cycle approval rates |
|
|
AF |
| 3. Products with Orphan status designation have higher first-cycle approval rates |
|
|
AF |
| 4. Products with fee waived have higher first-cycle approval rates |
|
|
AF |
| 5. Products with novel mechanism of action (MOA) have higher first-cycle approval rates |
|
|
ANF |
| 6. Products for indications classified as life-threatening or for unmet medical needs have greater first-cycle approval rates |
|
|
ANF |
| 7. Products that have a novel MOA and are for life-threatening conditions have higher first-cycle approval rate |
|
|
AF |
| 8. In-licensed drugs have greater approval rates |
|
|
ANF |
| 9. Novel MOA under In-licensed have greater approval rates |
|
|
ANF |
| 10. Products with significant public benefits are less likely to require multiple review cycles (treating life threatening disease?) |
|
|
NA |
| 11. Products for chronic conditions are more likely to require more than one review cycle |
|
|
ANF |
| 12. Products with more therapeutic areas involved are likely to require multi-cycle reviews |
|
|
NA |
| 13. Products addressing conditions with higher disease incidence are less likely to require multiple review cycles |
|
|
NA |
| 14. Products with international approval are less likely to require multiple review cycles |
|
|
ANF |
| 15. Applications with more Indications submitted are less likely to have single review cycle |
|
|
NA |
| 16. Increasing disease severity is likely to decrease the likelihood of multiple review cycles |
|
|
NA |
| 17. Products preceded by FDA approvals in same drug class are less likely to require multiple review cycles |
|
|
NA |
| 18. Products treating conditions with strong public advocacy are less likely to require multi-review cycles for approval (e.g., HIV/AIDS) |
|
|
NA |
| 19. NDAs are less likely to require multi-cycle reviews than BLAs |
|
|
AF |
| 20. Products that have secondary endpoints fail are more likely to require multiple review cycles |
|
|
NA |
| 21. Applications with international clinical trial sites will increase the likelihood of multiple review cycles |
|
|
NA |
| 22. Applications with more clinical trial sites increase the likelihood of multiple review cycles |
|
|
NA |
GRMPs Compliance
| Hypotheses | Metric(s) | Data Source | Status |
|---|---|---|---|
| 23. Implementing GRMPs will contribute to an increase in the first-cycle approval rate |
|
|
AF |
| 24. Product reviews that adopt all activities and timeframes in the first phase (filing and review planning) of GRMPs Guidance have greater first-cycle approval rate |
|
|
ANF |
| 25. Product reviews that adopt all activities and timeframes in the second phase (review) of GRMPs guidance have greater first-cycle approval rate |
|
|
ANF |
| 26. Product reviews that adopt all activities and timeframes in the third phase (Advisory Committee meeting) of GRMPs guidance have greater first-cycle approval rate |
|
|
ANF |
| 27. Product reviews that adopt all activities and timeframes in the fourth phase (Action) of GRMPs guidance have greater first-cycle approval rate |
|
|
ANF |
| 28. To be compliant with GRMPs timelines will increase initial workload |
|
|
NA |
| 29. Implementing GRMPs will result in earlier interactions between FDA and sponsor |
|
|
AF |
| 30. Implementing GRMPs will result in more frequent interactions between FDA and sponsor |
|
|
AF |
| 31. Implementing GRMPs will result in earlier interactions within the review team |
|
|
NA |
| 32. Implementing GRMPs will result in more frequent interactions within review team |
|
|
NA |
| 33. Implementing GRMPs will increase the review quality |
|
|
NA |
| 34. Implementing GRMPs will result in less compression towards the end of the review |
|
|
ANF |
| 35. Implementing GRMPs will increase the efficiency |
|
|
NA |
| 36. Implementing GRMPs will shorten review/ approval time |
|
|
NA |
| 37. Implementing GRMPs will increase the clarity of FDA expectations (internally and externally) |
|
|
ANF |
| 38. Implementing GRMPs will increase the transparency between FDA and sponsor |
|
|
ANF |
| 39. Implementing GRMPs will increase the consistency in review process |
|
|
ANF |
| 40. GRMPs training could increase the review efficiency |
|
|
NA |
| 41. Implementing GRMPs will increase the submission quality resulting in fewer IR letters and fewer filing or review issues |
|
|
ANF |
Issues and Communication
| Hypotheses | Metric(s) | Data Source | Status |
|---|---|---|---|
| 42. Sponsor response times to information requests correlate with first-cycle approval rate |
|
|
ANF |
| 43. Products in which the sponsor adequately prepares for the industry meetings are less likely to require multiple review cycles |
|
|
NA |
| 44. Early and effective interactions between FDA and Sponsor to identify, prioritize, and resolve issues early are likely to increase the first-cycle approval rate |
|
|
AF |
| 45. Products that have Pre-NDA/BLA meeting prior to submission are less likely to require multiple cycle reviews |
|
|
AF |
| 46. Products that have Pre-NDA/BLA meeting less than 6 months prior to submission are less likely to require multiple cycle reviews |
|
|
AF |
| 47. Products that have EOP2 meeting prior to submission are less likely to require multiple cycle reviews |
|
|
AF |
| 48. Products that have both EOP2 and Pre-NDA/BLA meetings prior to submission are less likely to require multiple cycle reviews |
|
|
AF |
| 49. Frequent communication is likely to increase the first-cycle approval rate |
|
|
AF |
| 50. Increased communication towards the end of the review cycle is likely to decrease the first-cycle approval rate |
|
|
AF |
| 51. Increasing number of issues identified in pre-submission meetings and correspondence are more likely to predict multiple review cycles (Core issues not the number of issues) |
|
|
AF |
| 52. Increasing number of issues identified during review are more likely to result in multiple review cycles (Core issues not the number of issues) |
|
|
AF |
| 53. Applications with increasing number of issues raised under the Trial Design phase are likely to increase the number of review cycles |
|
|
AF |
| 54. Reviews requiring Advisory Committee meeting are more likely to require multiple cycle reviews |
|
|
AF |
| 55. Types of Issues raised in Advisory Committee reviews may impact the number of review cycles required |
|
|
NA |
| 56. Necessity of more complex responses (e.g., new trial data) from FDA is more likely to result in multiple review cycles |
|
|
ANF |
| 57. Non-compliance of sponsor with specific FDA requests will increase the likelihood of multiple review cycles |
|
|
ANF |
| 58. FDA response to sponsor close to PDUFA goal date will result in multiple review cycles |
|
|
ANF |
| 59. Changes in submission requirements will increase likelihood of multiple review cycles. Such requirements include: endpoints, safety, efficacy and manufacturing standards |
|
|
NA |
| 60. Products with a greater number of unsolicited amendments are less likely to be approved first-cycle |
|
|
ANF |
| 61. Products with a change in the safety profile (as defined by FDA) for a given product class are less likely to be approved first-cycle |
|
|
NA |
| 62. Products submitted in eCTD or electronic NDA/BLA have greater first-cycle approval |
|
|
ANF |
| 63. Increased communication between FDA and the sponsor for products with Priority review designation leads to greater first-cycle approval |
|
|
AF |
| 64. Products with a clinical SPA review by FDA have greater first-cycle approval |
|
|
ANF |
| 65. Products with issues reported on the 74-Day letter that were discussed pre-submission are less likely to be approved first cycle |
|
|
ANF |
| 66. Products with any clinical or safety issue (not a data, data format, or label request) included in the 74-Day letter are less likely to be approved first cycle |
|
|
ANF |
| 67. Products with 74-Day letter issues that are not addressed during the review are less likely to be approved first cycle |
|
|
AF |
| 68. Products with unresolved safety and efficacy issues identified pre-submission (Pre-NDA/BLA, EOP2) are less likely to be approved first cycle |
|
|
ANF |
| 69. Products with more sponsor meetings during the review cycle are less likely to have multiple review cycles |
|
|
AF |
| 70. The use of checklists by reviewers increases the first-cycle approval rate |
|
|
ANF |
| 71. Divisions that have structured internal team meetings and documentation practices have a higher approval rate |
|
|
ANF |
| 72. High quality application (e.g., number of IRs, number of submission between application receipt date and 60-day filing date) submission have greater first-cycle approval rates |
|
|
ANF |
| 73. Large US-based companies are likely to have greater first-cycle approval rates |
|
|
AF |
| 74. Companies with more experience with FDA have greater first-cycle approval rates |
|
|
AF |
| 75. Companies with more experience in the therapeutic area have greater first-cycle approval rates |
|
|
AF |
| 76. Outsourcing consultant involvement is less likely to result in multiple review cycles |
|
|
ANF |
| 77. Sponsors whose prior submission characteristics shared with that of the current submission are less likely to require multiple review cycles |
|
|
NA |
| 78. Sponsors with experience in gaining application approvals since PDUFA are less likely to require multiple review cycles |
|
|
AF |
FDA Characteristics
| Hypotheses | Metric(s) | Data Source | Status |
|---|---|---|---|
| 79. Submissions received in the fourth quarter will have the lowest first-cycle approval rate |
|
|
AF |
| 80. Products that have facility inspections early in the review cycle are more likely to increase first-cycle approval rates |
|
|
NA |
| 81. Applications requiring foreign facility inspection have lower first-cycle approval rates |
|
|
AF |
| 82. Reviewer or Division workload is likely to contribute to multi-cycle review |
|
|
NA |
| 83. Applications that experience turnover of lead reviewers (i.e., Office Director, Clinical Team Lead) are more likely to result in multiple review cycles |
|
|
AF |
| 84. Application that experience turnover of the RPM are more likely to result in multiple review cycles |
|
|
AF |
| 85. Applications that are submitted to a Division in which the Division Director changes during application review result in more multi-cycle reviews |
|
|
AF |
| 86. Applications that have more postmarketing commitment studies are less likely to require multiple review cycles |
|
|
AF |
| 87. Drug-Device combinations requiring input from multiple divisions or areas are more likely to require multiple cycles |
|
|
NA |
| 88. Drug-Drug combinations requiring input from multiple divisions or areas are more likely to require multiple cycles |
|
|
NA |
| 89. Increasing numbers of consults (internal and external) are more likely to result in multiple review cycles |
|
|
ANF |
| 90. Type and timing of consult (internal vs. external) may impact the number of review cycles required |
|
|
ANF |
| 91. Number and type of issues raised in consults may impact the number of review cycles required |
|
|
NA |
| 92. Applications with dissenting opinions on the Priority review designation amongst reviewers are more likely to require multiple review cycles |
|
|
NA |
| 93. An experienced reviewer (2+ years of regulatory review) will have higher first-cycle approval rates |
|
|
NA |
*Approval Rate metric will be based on specific hypothesis (e.g., for hypothesis related to the Fast-Track Program, the approval rate measured will be for approvals within this program category)
[1] Independent Evaluation of FDA's First Cycle Review Performance – Retrospective Analysis Final Report, Booz Allen Hamilton Inc., January 2006 http://www.fda.gov/ope/pdufa/PDUFA1stCycle/default.htm
[2] For the purposes of evaluating GRMPs compliance and impact, five key activities and associated timelines were considered based on the importance of the activity, as well as the availability of information to assess compliance with these activities: hold filing meeting, communicate filing review issues to applicant, hold mid-cycle meeting, complete primary review, and hold labeling discussions (for approval and approvable actions).
[3] Review issues are those issues identified during the filing of an application that may potentially impact approval.
[4] Action packages are a collection of important documents generated during the review of an application.
[5] The PDUFA III goals can be found at http://www.fda.gov/oc/pdufa/PDUFAIIIGoals.html. As of September 2007, FDA began operating under PDUFA IV. PDUFA IV goals can be found at http://www.fda.gov/oc/pdufa4/pdufa4goals.html
[6] Independent Evaluation of FDA's First Cycle Review Performance – Retrospective Analysis Final Report, Booz Allen Hamilton Inc., Jan. 2006 http://www.fda.gov/ope/pdufa/PDUFA1stCycle/default.htm
[7] Six of the 185 applications did not reach action until January 2008
[8] The 77 products analyzed in “The Independent Evaluation of FDA’s First Cycle Review Performance – Retrospective Analysis Final Report” are included in the Overall Study Cohort.
[9] A user fee waiver may be granted for a small business submitting its first application. Also, a waiver may be granted where: it is necessary to protect the public health; assessment of the user fees would present a significant barrier to innovation due to limited resources; fees will exceed the anticipated costs incurred by FDA for conducting the application review; assessment of the fee for an application filed under section 505(b)(1) pertaining to a drug product would be inequitable because an application for a product containing the same active ingredient filed by another person under section 505(b)(2) could not be assessed user fees. (Section 736(d) of the Federal Food, Drug & Cosmetic Act (FDCA))
[10] Guidance for Review Staff and Industry: Good Review Management Principles and Practices for PDUFA products. April 2005.
[11] Only the FY2005-FY2007 cohort could be analyzed, since the data source for earlier products was limited to Action Packages, which contain limited if any information regarding pre-submission meetings.
[12] SOPP 8401.3. Filing Action – Communication Options. Version #1. May 11, 2003
[13] PDUFA III goals were to provide the sponsor a notification of deficiencies prior to the goal date for 50% of applications in FY 2003, 70% in FY 2004, and 90% in FY 2005, FY2006, and FY 2007.
[14] Formal Meetings with Sponsors and Applicants for PDUFA Products Guidance
[15] A major deficiency is defined as a product- or application-related issue that would contribute to preventing first-cycle approval if not adequately addressed.
[16] Issues identified in either the EOP2 or Pre-NDA/BLA meeting are grouped in one pre-submission category because sponsors can take as much time as needed to resolve an issue before submitting the application, but must resolve them by the Action Date for issues identified during the review.
[17] Title IX, Subtitle A, Section 901 of the Food and Drug Administration Amendments Act of 2007 (FDAAA) amends the FDCA to authorize FDA to require holders of approved drug and biological product applications to conduct postmarketing studies and clinical trials for certain purposes (section 505(o)(3)(A), 21 U.S.C. 355(o)(3)(A)). This provision took effect on March 25, 2008. The description in this report refers to policies in place prior to FDAAA implementation.
[18] Analysis not shown.
[19] Pre-submission documentation of RPM and Medical team lead were not available for all products.
[20] Contributing factor was determined by reviewing deficiencies noted on the first-cycle Action Letter.
[21] Review teams that performed each activity within one week of the guideline were considered fully compliant. “Highly compliant” review teams performed at least 4 out of the 5 activities within one week of the guideline (80% compliant).
[22] The activities include: hold filing meeting, communicate filing review issues to applicant, mid-cycle meeting, complete primary review, and labeling discussions (for approval and approvable actions).
[23] Note: Large pharmaceutical companies had a market capitalization over $5B; large biotechnology had capitalization over $1B; small biotechnology had capitalization under $1B, small-medium pharma had less than $5B.
[24] At the Application Orientation meeting, sponsors can walk the FDA through the format of their product application. However, discussion of data, results, and conclusions is not the focus of this meeting.
![]()