 |
 |
 |
FDA
Home Page | Search FDA Site |
FDA A-Z Index | Contact FDA
![]()
Printer-friendly PDF [4.28 MB]
The Prescription Drug User Fee Act (PDUFA) program is the cornerstone of modern FDA drug review. User fees currently fund about half of new drug review costs. By providing needed funds, PDUFA ended slow and unpredictable review and approval of new drug applications, while keeping FDA's high standards.
PDUFA funds allowed FDA to accomplish a number of important goals. FDA hired more review and support staff to speed review. The number of full-time equivalent (FTE) staff devoted to the new drug review process has nearly doubled, growing from 1,277 FTE in 1992 to 2,503 FTE in 2004. FDA upgraded its data systems and gave industry guidance to help minimize unnecessary trials and generally improve drug development. FDA gave industry guidance on how to improve the quality of applications, with the goal to reduce misunderstandings and the need for sponsors to rework and resubmit applications. Finally, FDA improved procedures and standards to make review more rigorous, consistent, and predictable. [Section 1]
Taken together, all of these steps ensure that the time and effort patients put into clinical trials provide useful data. They also lowered drug development costs and shortened review times. For example, the median approval time for priority new drug applications and biologics license applications decreased from 13.2 months in 1993 to 6.4 months in 2003. Ultimately, these developments enabled FDA to ensure that needy American patients get fast access to novel drugs--faster, in fact, than citizens of other countries. Since the start of PDUFA, FDA has approved over 1,000 new drugs and about 100 new biologics. Under the currently authorized program (PDUFA 3) 50 percent of new drugs are launched first in the United States, compared to only 8 percent in the years pre-PDUFA.
To get to these outcomes, PDUFA established goals acceptable to FDA and new drug sponsors for both reviewing drugs and giving industry guidance during drug development. These goals included the following. [Section 3]
Additional goals specifically focused on preserving an appropriate balance between drug efficacy and drug safety by funding safety-related activities for the first 2 years of product marketing for most drugs, and the first 3 years for potentially dangerous drugs. PDUFA fees also enabled FDA to issue guidance for FDA and industry on how best to assess, manage, and monitor drug risk. [Sections 3.4-3.5]
However, since the PDUFA program began, FDA's drug review workload substantially increased. [Section 4]
At the same time that workload has increased, FDA's capacity to deliver timely and predictable performance is challenged by cost pressures, budget constraints and the need to meet other important work mandates. These challenges have been accompanied by flat to negative growth of total appropriated funds to FDA. The challenges include: mandated federal pay increases and earmarks for important non-PDUFA programs including activities around generic drugs, blood safety, and influenza vaccine; the costs of recruiting and retaining qualified expert (marketable) reviewers; and normal market-driven increases in the cost of facility upkeep and rent for necessary work space. [Section 5]
The Prescription Drug User Fee Act (PDUFA), which authorizes FDA to collect user fees amounting to a little more than half of total funding for drug review, expires in September 2007. Without further legislation, FDA would be unable to collect user fees for the new prescription drug review program. This paper provides data and information on the scope and current status of FDA's program to implement PDUFA, so as to inform public discussions regarding PDUFA reauthorization.
PDUFA has had a significant role in modern drug review at FDA. It has been a key to ending major problems with unpredictable and slow review and approval of new drug applications. It has provided funds to eliminate or even reversed the so-called "drug lag" attributed to inadequate staff and computer resources. Americans now get access to more new medicines faster than patients in other countries, while prior to PDUFA, American patients waited for FDA to act long after new drugs were available in Europe.
Reauthorization of PDUFA in 1997 (PDUFA 2) and 2002 (PDUFA 3) established a precedent for a process to identify areas to consider related to PDUFA 4 in 2007. FDA seeks public comment on the current program, including views on what features should be retained and what might be done to further strengthen and improve the program. On November 14, 2005, FDA will hold a public meeting on PDUFA and will at the same time open a public docket where people can submit written comments. In addition, per provisions of the statute, the Secretary of DHHS will consult with the Committee on Energy and Commerce of the House of Representatives, the Committee on Health, Education, Labor, and Pensions of the Senate, appropriate scientific and academic experts, health care professionals, representatives of patient and consumer advocacy groups, and the regulated industry. The Secretary will publish in the Federal Register any recommendations after negotiations with the regulated industry and will present the recommendations to the congressional committees. In addition, the Secretary will publish a Federal Register notice including department recommendations to Congress regarding PDUFA reauthorization. Following that FR notice, FDA will hold another public meeting to hold a meeting at which the public may present its views on those recommendations; and will provide for a period of 30 days for the public to provide written comments on those recommendations.
This white paper summarizes major factors behind establishment of the program. The paper describes how the fees are collected and what they pay for, and how the PDUFA program has evolved since its enactment in 1992. In addition, the paper describes progress to date, and current challenges for the funding of drug review and achieving the related public health aims.
Drug review at FDA prior to PDUFA entailed a variety of problems that PDUFA alleviated. Before PDUFA, FDA's review process was understaffed, unpredictable and slow. FDA lacked sufficient staff to perform timely reviews, or develop procedures and standards to make the process more rigorous, consistent and predictable. FDA lacked the funds to provide computers to all FDA reviewers. At the same time, regulators in other countries were able to review products faster. Access to new medicines for U.S. patients lagged behind. For example, U.S. patients were not getting access to new AIDS drugs as quickly as in other countries. Chronic understaffing of drug review, and related delays in U.S. patient access to new drugs led to the 1992 enactment of PDUFA.
PDUFA was established to increase FDA funding for human drug review and to increase the speed and predictability of the review process. Under PDUFA the U.S. pharmaceutical and biotechnology industries would pay user fees and FDA would commit to develop a more standardized process and faster, more predictable review timeframes. (Section 4 provides more details on the performance goals related to these commitments.) PDUFA provided FDA with added funds that enabled the agency to hire additional reviewers and support staff and upgrade its information technology systems to speed up the application review process for new drugs and biological products without compromising FDA's high standards for approval.
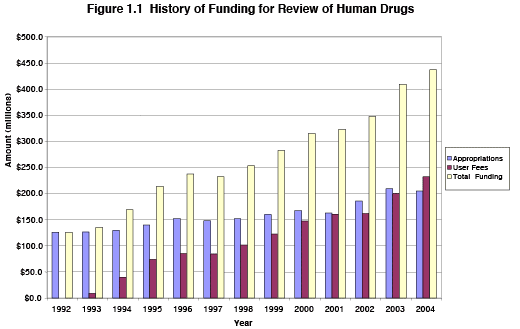
Since the beginning of the program, there has been a significant improvement in FDA funding for the drug review program, including significant investments in information technology. As shown in Figure 1.1, PDUFA has enabled the nominal funding for human drug review to increase by over 225 percent from 1992 to 2004. Because Congressional appropriations have grown at a much lower rate, user fees now represent over half of all resources devoted to the review of human drugs.
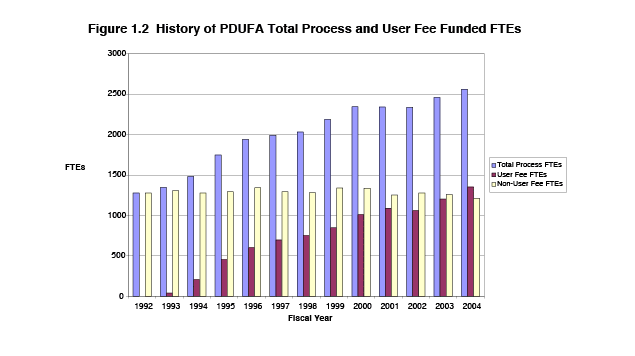
PDUFA has resulted in a significant increase in staffing, enabling FDA to provide more guidance for industry during drug development and improvements to the review process. As shown in Figure 1.2, the number of full-time equivalent staff (FTEs) devoted to the new drug review process nearly doubled between 1992 and 2004, increasing from 1,277 FTEs in 1992 to 2,503 in 2004. The increased share of program support from user fees means that currently more than half of all FTEs devoted to the review of human drugs are now funded by user fee revenues.
FDA committed to certain performance goals on timeframes for the review of new drug applications (NDAs) and biologics license applications (BLAs). FDA has met or exceeded all PDUFA NDA and BLA review goals since PDUFA's inception.
However, meeting review goals is not the only desired outcome. By meeting the review goals, FDA has been able to dramatically reduce the time to approval for new drugs and biologics and provide the American public with access to these medical treatments more quickly.
Since the start of PDUFA, FDA has approved 1,010 new drug applications and about 100 biologics licensing applications. These include 62 new cancer drugs, 109 new drugs for metabolic and endocrine disorders, 96 new anti-infective drugs, 103 new drugs to treat neurologic and psychiatric disorders and 73 new drugs to treat cardiovascular and renal disease.
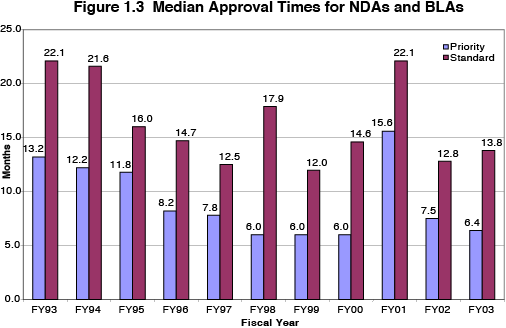
Between 1993 and 2003 the median approval time for priority NDAs and BLAs decreased by over half -- from 13.2 months in 1993 to 6.4 months in 2003. Over this same time period the median approval time for standard NDAs and BLAs decreased by over one third, from 22.1 months in 1993 to 13.8 months in 2003. Figure 1.3 shows the median approval times for NDAs and BLAs by priority status for the PDUFA era, and depicts the overall trend toward shorter times to new drug approval for both priority and standard drug applications.
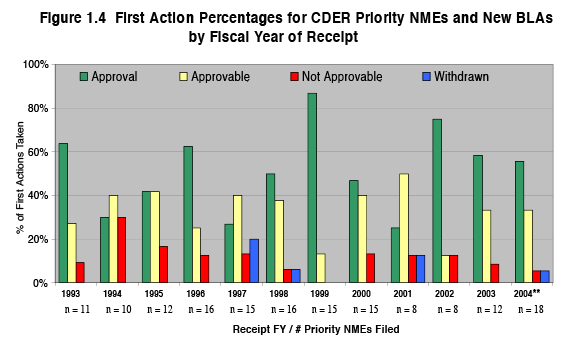
The reduction in median approval times has resulted from significant improvements in FDA review process efficiencies, while holding to the same high standard of evidence for drug safety and effectiveness. This is evident in the data showing the percentage of applications approved on the first cycle [i.e., the first time they are submitted for FDA review] versus approved on a subsequent review cycle after they have successfully addressed deficiencies identified by FDA in a previous review. As shown in Figures 1.4 the percentage of priority applications approved on first cycle has changed very little over the years, even as the median approval times have declined.
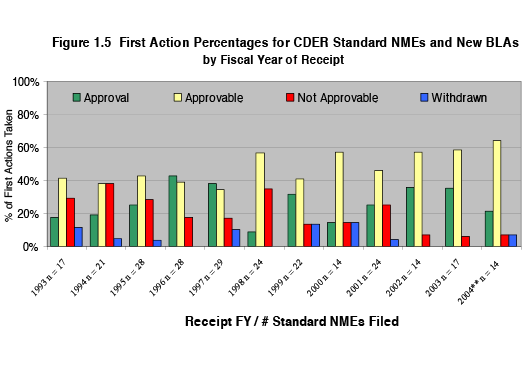
The data for percentage of first versus later-cycle approvals for standard drug applications also suggest that there is no trend toward increasing approvals in the first cycle. However, as shown in Figure 1.5, the data do suggest that an increasing percentage of sponsors are effectively addressing deficiencies identified in the first cycle, and are able to obtain approval after the second cycle of review.
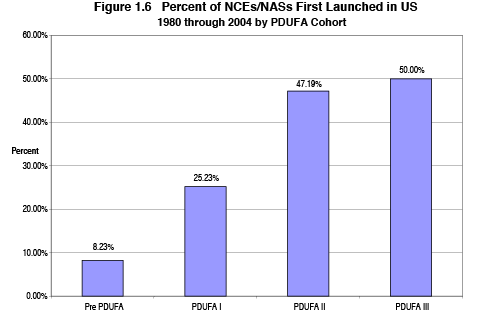
The combination of realistic time frames, management improvements, and additional resources improved FDA's review performance for drugs and biologics. Since the start of PDUFA, the so-called drug lag in America, compared to other countries, has been reversed and the proportion of all new drugs first marketed in the United States has increased dramatically. Figure 1.6 shows the percent of new chemical entities (NCEs)/new active substances (NASs) first launched in the United States: pre-PDUFA period of 1980-1992, PDUFA 1 years 1993-1997, PDUFA 2 years 1998-2002 and PDUFA 3 2003-2004.
The design of PDUFA user fees detailed in the provisions of the statute, has contributed to the program's stability and success. This section reports on the design and the current level of fee collections.
PDUFA fees are collected in three components: application fees, establishment fees, and product fees. Each of the three types of fees contributes one third of the total revenues in a fiscal year. An application fee must be submitted when certain new drug applications (NDAs) or biologic license applications (BLAs) are submitted. Product and establishment fees are due annually on October 1. The total revenue amounts derived from each of the categories--application fees, product fees, and establishment fees--are set by the statute for each fiscal year. The following schedule was included in PDUFA 3:
| Type of Fee Revenue | Fiscal Year 2003 | Fiscal Year 2004 | Fiscal Year 2005 | Fiscal Year 2006 | Fiscal Year 2007 |
|---|---|---|---|---|---|
| Application/Supplement | $74,300,000 | $77,000,000 | $84,000,000 | $86,434,000 | $86,434,000 |
| Establishment | $74,300,000 | $77,000,000 | $84,000,000 | $86,433,000 | $86,433,000 |
| Product | $74,300,000 | $77,000,000 | $84,000,000 | $86,433,000 | $86,433,000 |
| Total Fee Revenue | $222,900,000 | $231,000,000 | $252,000,000 | $259,300,000 | $259,300,000 |
The increase in revenues each year was provided to give FDA sufficient resources to meet the performance goals that are also agreed to over the 5 years of the program.
PDUFA authorizes adjustments in fees to account for cost inflation and workload changes. The statutory amounts shown in the box above must be adjusted for cumulative inflation since FY 2003 and for changes in drug review workload in each fiscal year. PDUFA 3 authorizes FDA in each fiscal year to set user fees so the total revenue that FDA receives from each fee category approximates the statutory amounts, after the adjustments for inflation and the workload. Those fees, with the rational for their calculation, are published in the Federal Register each year in early August before the beginning of the Fiscal year on October 1. The fees for the first 4 years of PDUFA 3 are shown in the table below.
| Type of Fee Revenue | FY 2003 | FY 2004 | FY 2005 | FY 2006 |
|---|---|---|---|---|
| Application Fee | $533,400 | $573,500 | $672,000 | $767,400 |
| Establishment Fee | $209,900 | $226,800 | $262,200 | $264,000 |
| Product Fee | $32,400 | $36,080 | $41,710 | $42,130 |
The fees for applications have risen faster than the other fees because the estimate of the number of full applications fees that FDA will receive, and that will produce the revenue expected, has gone down from an estimated 139 in 2003 to an estimated 129 in 2006. (It should be noted that about 25 percent of all applications received each year do not pay fees because the applications are for fee-exempt orphan products, or the fees are waived either because it is the first application from an small business or it qualifies for one of the other waiver provisions. The estimates of the numbers of fees that will be received is based on a rolling average of the number of full application fees received in the previous 5 fiscal years.)
PDUFA has provisions intended to ensure that the resources from user fees are in addition to and not instead of Congressional appropriations to support the process of human drug review. It imposes three legal conditions on FDA's authority to collect the user fees.
So far, FDA has been able to meet these three conditions.
FDA administration of fee collections is designed to ensure both efficiency and complete separation of fee collections from fee-supported review activities. The collection of fees for applications is done through Mellon Bank and coordinated by FDA's Office of Financial Management in the FDA Office of Commissioner. The annual bills for establishment fees and product fees are based on FDA's database of listed products and active establishments, and fee monies are then collected centrally by Mellon Bank. FDA drug reviewers and other staff are generally unaware of whether and how much any individual company has paid in user fees. The table below summarizes fee collections for the most recently available Fiscal Years 2003 and 2004.
As of September 30, 2004
| Fee Category Fees Collected: |
FY 2003 | FY 2004 |
|---|---|---|
| Product Fees | $76,852,785 | $76,453,520 |
| Establishment Fees | $78,209,219 | $82,318,894 |
| Application Fees | $62,684,550 | $87,693,991 |
| Total Fees Collected: | $217,746,554 | $246,466,405 |
FDA spends user fee revenues only on activities to support the "process for the review of human drug applications"[1], as defined in PDUFA 3. Under PDUFA, fees collected and appropriated, but not spent by the end of a fiscal year, continue to remain available for FDA to spend in future fiscal years. In FY 2004, FDA obligated $232,081,500 from user fee revenues. Those costs were distributed as shown in the table below. The amounts are based upon the obligations recorded as of the end of each fiscal year. In the past, over 81 percent of amounts obligated are expended within one year, and 96 percent within two years. Thus, annual obligations represent an accurate measure of annual costs.
As of September 30, 2004
| FDA Component | FY 2003 | FY 2004 |
|---|---|---|
| Center for Drug Evaluation and Research (CDER) | $250,370,170 | $293,991,408 |
| Center for Biologics Evaluation and Research (CBER) | $110,132,866 | $91,905,443 |
| Field Inspection and Investigation Costs (ORA) | $19,098,382 | $19,646,087 |
| Agency General and Administrative Costs (OC) | $29,840,492 | $31,313,598 |
| Total Process Cost | $409,441,910 | $436,856,536 |
|---|---|---|
| Amount from Appropriations | $$209,287,410 | $$204,775,036 |
| Amount from Fees | $200,154,500 | $232,081,500 |
The costs for all components, except for CBER, rose in FY 2004. The increased expenditures primarily reflect the additional personnel hired by the organizations in FY 2004 and the mandatory pay raise for all federal employees. The reason for the decrease in CBER and the increase in CDER is because of the transfer of review responsibility for certain therapeutic biologics from CBER to CDER in FY 2004. The methodology used to develop the estimated full cost of human drug review is detailed in Appendix D of the FY 2004 PDUFA Financial Report.
The activities encompassed in the PDUFA "process of human drug review," that are needed to improve drug development and speed patient access, begin well before a product sponsor submits a new drug application for FDA pre-market review. Although not often discussed, these other--often FDA labor-intensive--activities are critical to improving the quality of a sponsor's drug development and the quality of a submitted market application. High-quality development is crucial to patient safety during clinical trials and to ultimate approval of a safe and effective product. Application quality is a decisive factor in drug review and approval.
PDUFA funding has enabled increased review staffing to increase FDA-sponsor interactions for scientific and regulatory consultation at a number of critical milestones in drug development, as shown in Figure 3.1.
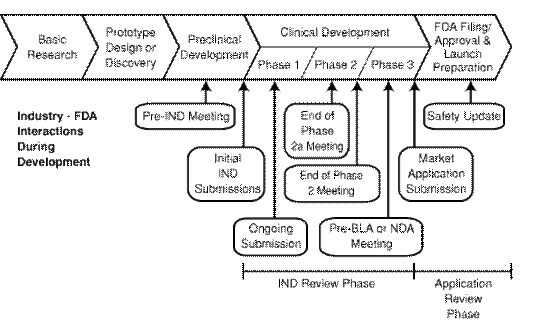
This section of the paper provides more background information about FDA activities from consultation in preclinical development through review of submitted risk management plans and safety surveillance following new drug approval. At the end of each subsection is a brief summary of current requirements under PDUFA 3.
For truly new therapies, sponsors often request a meeting with FDA to discuss their preclinical findings and review their plans for clinical development. During the preclinical phase of drug development, sponsors' primary goal is to determine whether: 1) the product is reasonably safe for initial use in humans and 2) it is sufficiently effective against a disease target in chemical assay tests or in animal models to justify the cost of commercial development. When a product is identified as a viable candidate, the drug's sponsor then focuses on demonstrating its effectiveness and on collecting the safety data and dosing information necessary to establish that the product will not expose humans to unreasonable risks when used in limited, early-stage clinical studies in humans.
The clinical phase of product development (see Figure 3.1) extends from a sponsor's initial submission of the Investigational New Drug Application, to begin testing a new drug in humans (i.e., clinical studies) to submission of a complete NDA)or BLA to FDA for marketing approval.
The IND application must contain information in three broad areas:
Once the initial IND is submitted, the sponsor must wait 30 calendar days before initiating any clinical trials. During this time, FDA has an opportunity to review the IND for safety and compliance (the toxicology data and the clinical study protocol) to ensure that research subjects will not be subjected to unreasonable risk. When reviewing investigational new drug applications, FDA can decide whether it is reasonably safe for the sponsor to move forward with testing the drug on humans; if FDA concludes trials cannot proceed safely, it puts the IND on clinical hold.
Under PDUFA, FDA's goal is to reply to a sponsor's complete response to a clinical hold within 30 days of the Agency's receipt of the submission of such sponsor response, and do this for at least 90 percent of such submissions. Rapid resolution of safety issues that led to clinical hold helps ensure patient safety while enabling access to the experimental treatment.
Clinical trials conducted to obtain evidence of the safety and effectiveness of a new drug are described in terms of three successive phases. The table below provides a brief summary.
Phase 1Number of Patients: 20-100 |
Phase 2Number of Patients: Up to several hundred |
Phase 3Number of Patients: Several hundred to several thousand |
Phase 1 Studies--begin if FDA does not impose a clinical hold. The focus is primarily on safety, with the goal of determining the relationship between dosing and the patient's systemic drug exposure; the drug's most frequent side effects and whether they are related to dose; and how the drug is metabolized and excreted.
Phase 2 Studies--begin if Phase 1 studies do not reveal unacceptable toxicity. The emphasis is primarily on effectiveness and collection of preliminary data on whether the drug works in people who have a certain disease, and the relationship between dose and effectiveness. For controlled trials, patients receiving the drug are compared with similar patients receiving a different treatment -- usually a placebo or a different drug. Safety continues to be evaluated, and short-term side effects are studied.
Phase 3 Studies--begins if preliminary evidence of effectiveness is shown during phase 2. The goal is to gather more information about safety and effectiveness, about effects on different populations, and about different dosages. Data are also gathered on how the drug interacts when used in combination with other drugs (drug-drug interactions).
Under PDUFA, FDA will evaluate specific questions about the sponsor's special study protocol designs for carcinogenicity, stability and Phase 3 for clinical trials that will form the primary basis of an efficacy claim.
This FDA review and written feedback ensures safer and more effective study design for participating patients and increases likelihood that resulting marketing application will meet regulatory requirements and gain faster approval.
Sponsors usually request to meet with FDA during their product's clinical development. The two most common meeting points are at the end of phase 2 clinical trials and just before a new drug application (NDA) or a biologic license application (BLA) is submitted to FDA for marketing review.
End of Phase 2 Meeting--in which sponsors seek FDA input on their Phase 3 clinical studies that are generally pivotal to FDA application review and market approval. Phase 3 studies are designed to demonstrate effectiveness in accordance with FDA written guidance or individually developed (e.g., drug or disease specific) study expectations, the duration of effect, and the populations that will be studied. The studies are also designed to provide adequate assessment of safety, including concerns raised by other members of the drug class or by phase 2 observations.
Pre-NDA/BLA Meeting--in which sponsors seek to discuss what FDA expects to see included in the submitted NDA or BLA application. The discussion between the FDA and the sponsor would generally address concerns raised during clinical studies and the limitations of those studies and planned risk management tools (labeling and others) to address known and potential risks. The discussion will include suggestions for phase 4 studies, if such studies are warranted. In addition, the meeting will consider any proposals for targeted post-approval surveillance, including attempts to quantify background rates of risks of concern and thresholds for actions. The intent of these discussions will be for FDA to get a better understanding of the safety issues associated with the particular drug and any proposed risk management plans, and to provide industry with feedback on these proposals so that they can be developed fully and included in the NDA submission, if needed.
Under PDUFA, FDA's goal is to notify the sponsor in writing, of the date, time, place and FDA participants for a formal meeting within 14 calendar days of the sponsor's request. FDA's goal is to meet the following timeframes for 90 percent of requested meetings:
Earlier consultation and feedback from FDA on the sponsor's development program will improve the quality of development including safer and more effective study design for patients and increases the likelihood that resulting marketing application will meet regulatory requirements and gain faster approval.
If a sponsor successfully completes the clinical development phase for a new drug, the next step is to submit an NDA or BLA to FDA. Since 1938, every new drug has been the subject of an approved NDA before it could be sold in the United States. An NDA (or BLA) includes all animal and human data and analyses of that data, as well as information about how the drug behaves in the body and how it is manufactured. The data gathered during the animal studies and human clinical trials of an IND application become part of the NDA. Under law, no pertinent data may be omitted.
When an NDA or BLA is submitted, FDA has 60 days to determine whether the application is complete enough to file and be reviewed. The FDA can refuse to file an application that is incomplete (e.g., required studies are missing). Once the application is filed, the review schedule begins, and FDA monitors the progress of the application over time. FDA expects to review and act on at least 90 percent of NDAs for standard drugs and biologics no later than 10 months after the applications were filed. The review goal is 6 months for priority drugs and biologics. [2]
The NDA/BLA submission should provide enough information to permit FDA reviewers to reach the following critical decisions:
Once an application is filed, an FDA review team -- medical doctors, clinical pharmacologists, chemists, statisticians, microbiologists, pharmacologists and other experts (see box below) -- evaluates whether the studies the sponsor submitted show that the drug is effective for its proposed use and has been shown, on the basis of an adequate safety assessment, to have an adverse effect profile that allows a conclusion that the drug is safe for its intended use at the doses proposed by the sponsor. Because no drug is absolutely safe, "safe" in this sense means that the benefits of the drug appear to outweigh its risks.
Chemists focus on how the drug is made and whether the manufacturing process and packaging are adequate to ensure the identity, strength, quality, and purity of the product.
Pharmacologists and toxicologists evaluate the effects of the drug on laboratory animals in short-term and long-term studies.
Physicians evaluate the results of the clinical tests, including the drug's adverse as well as therapeutic effects, and whether the proposed labeling accurately reflects the effects of the drug.
Clinical pharmacologists evaluate the rate and extent to which the drug's active ingredient is made available to the body and the way it is distributed, metabolized and eliminated.
Statisticians evaluate the designs for each controlled study and the analyses and conclusions for safety and effectiveness based on the study data.
Microbiologists also participate in the review of anti-infective drug products and of products that occur as solutions or as injectables. For new biological products, the microbiologist may also be a product specialist focusing on how the biologic is manufactured and packaged to ensure potency and purity.
The review team analyzes study results and looks for possible problems with the application, such as weaknesses in the study design or analyses, and missing information that may be critical to determining drug safety. Reviewers determine whether they agree with the sponsor's results and conclusions, or whether they need any additional information to make a decision. Just as critical as the assessment of observed side effects is the determination of whether the safety assessment was of adequate scope. Each reviewer prepares a written evaluation containing conclusions and recommendations about the application. These evaluations are then considered by team leaders, division directors, and office directors, depending on the type of application.
In the course of its review, FDA may also call on advisory committees, made up of outside experts who make recommendations to FDA. Whether an advisory committee is called on depends on a number of things; for example, whether a drug raises significant safety questions, whether it is the first in its class, or is the first for a given indication.
If the drug presents a significant documented risk, and sometimes even if the concerns about such risks are theoretical, the NDA submitted by the sponsor may include proposed risk management tools, plans, and protocols for further studies. Both the FDA pre-market review and post-marketing surveillance staff are involved in the review of the risk management plan. When such a plan is needed, the risk management plan will be part of the overall safety and risk-benefit analysis. In some cases, FDA may seek a commitment from the sponsor for the conduct of specific studies after the drug is approved (phase 4 studies).
After a BLA is approved for a biological product, the product may also be subject to official lot release. If the product is subject to official release by FDA, the manufacturer submits samples of each lot of product to FDA. In addition, FDA conducts laboratory research related to the regulatory standards on the safety, purity, potency, and effectiveness of biological products.
Under PDUFA, FDA commits to perform a complete application review and will issue an action letter within a specified timeframe. The action letter will either state that the product is approved for marketing, is designated "not approvable, or the product is designated "approvable" if the sponsor can address the deficiencies detailed in the action letter. Sponsors receiving an approvable letter will often try to address the deficiencies and will then resubmit their application.
Under PDUFA FDA's goal is to review all filed original NDA/BLA submissions within the following time frames:
For all NDA/BLA resubmissions,
Rapid and complete review of new drug applications ensures rapid access to new medicines shown to be safe and effective; with priority given to products with greatest public health benefit. It also ensures complete information for sponsors to do the additional work needed to show that their product is safe and effective.
Following approval of an original NDA or BLA, a sponsor may later submit an application to expand the disease indications included in the drug labeling. This application often includes additional clinical study data to support the proposed change in labeling, and is referred to as an Efficacy Supplement. FDA's review of Efficacy Supplements is very similar to the review of original NDAs and BLAs, and results in an action letter to the sponsor outlining FDA's decision.
Under PDUFA FDA's goal is to review all filed original Efficacy Supplements within the following timeframes.
Rapid and complete review of new drug applications ensures rapid access to new medicines shown to be safe and effective; with priority given to products with greatest public health benefit. It also ensures complete information for sponsors to do the additional work needed to show that their product is safe and effective.
After a new drug has been approved for marketing, the sponsor begins mass production for commercial distribution of the product. The sponsor also begins marketing activities including detailing to physicians[3] by the drug company sales force and direct advertising to health professionals and often to consumers. The manufacturing and distribution must comply with the methods approved as part of the NDA, and drug promotion must also conform with and reflect the information in the FDA-approved labeling for the new drug.
Because all possible side effects of a drug cannot be anticipated based on preapproval studies involving only several hundred to several thousand patients, FDA maintains a system of post-marketing surveillance and risk assessment programs to identify adverse events that did not appear during the drug approval process.
Knowledge about a product will always be limited to some extent at the time of approval by factors in the product development process. For example, rare side effects and long-term outcomes (both positive and negative) may not be known when a product is approved because of the relatively small size and limited duration of clinical studies. And because of the populations not studied in clinical trials (e.g., pregnant patients, children, people with other diseases) or minimally studied (e.g., geriatric patients), side effects may be discovered if these groups are treated with a product after it goes on the market. Even after a product has been on the market for a long time, uncertainties remain. The Adverse Event Reporting System (AERS) provides FDA's primary source of post-marketing safety data.
New information about a new drug will also emerge from additional clinical trials, such as post-Marketing Commitments (PMCs, also known as Phase 4 studies), and from reports in the literature. FDA uses all available post-marketing safety information to investigate product quality problems, to update drug labeling, and, on more rare occasions, to reevaluate the approval or marketing decision to revise other aspects of risk management plans.
Recent public attention to the issue of drug safety has included questions about the extent of FDA oversight and the resources devoted to this critical aspect of public health protection. The most readily available numbers are usually budget figures for individual offices within FDA but the work done by those offices represents only a subset of the larger effort required to ensure drug safety.
To provide a more complete and accurate account of the full range of critical drug safety-related activities, FDA recently conducted an internal study, including extensive staff interviewing and analysis of staff time reporting data across CDER. The results of this analysis, based on FY 2004 data, suggest that CDER staff devote 50 percent of their time to drug safety activities. This represents a level of effort totaling about 1,093 full time equivalent (FTE) staff. These figures are based on the agency's FY 2004 staffing.
How does this level of effort compare to the total staffing for work performed in other FDA regulatory work? The table below provides a summary based on FY 2004.
| FDA Program Area | FY 2004 Actual - Total FTEs |
|---|---|
| Center for Food and Applied Nutrition | 910 |
| Center for Drug Evaluation and Research Level of Effort devoted to drug safety activities |
2,1901,093 |
| Center for Biologics Evaluation and Research | 797 |
| Center for Veterinary Medicine | 349 |
| Center for Devices and Radiologic Health | 1,061 |
| National Center for Toxicologic Research | 207 |
| Field Activities Program (Office of Regulatory Affairs) | 3,872 |
| Other Activities (including Office of Commissioner) | 755 |
| TOTAL | 10,141 |
PDUFA fee revenues enabled FDA to significantly increase the scientific review staffing in almost every CDER office involved in assuring drug safety. For example, since the beginning of PDUFA the additional funds have enabled CDER to more than double its medical staff, growing from 166 medical officers in 1993 to 342 medical officers on staff in 2004. This translates into significantly higher staffing to focus on drug safety issues.
In both premarket and postmarket phases, staff across CDER work to assure the highest possible degree of drug safety. In total, NDA review involves chemists, pharmacologists, toxicologists, physicians, statisticians, epidemiologists, pharmacists, biologists, microbiologists, project managers, health and social scientists. The Office of New Drugs (OND) leads and coordinates this review. Postmarket review requires similar varieties of FDA staff expertise found in various CDER Offices.
Safety Work in Specific CDER Offices.
| Office | Office Percent of Work Time | Premarket Estimated FTE |
Postmarket Estimated FTE | Total Office Estimated FTE |
|---|---|---|---|---|
| OND | 51% | 275 | 77 | 352 |
| OPaSS | 60% | 32 | 93 | 126 |
| [ODS] | [100%] | [9] | [87] | [96] |
| OMP | 44% | 6 | 29 | 36 |
| OCPB | 44% | 41 | 1 | 42 |
| OPS | 51% | 179 | 74 | 252 |
| OC | 38% | 13 | 30 | 43 |
| OCTaP | 73% | 22 | 13 | 35 |
| OEP | 14% | 4 | 4 | 8 |
| Mgt/Other | -- | 1282 | 712 | 1992 |
| Total CDER | 50% | 700 | 393 | 1,093 |
1 All estimates represent general levels of magnitude and are primarily for comparison purposes. 2 To get FTE estimates for "Mgt/Other" we applied overall CDER estimates of percent safety activity work time to offices and programs not included in the study. |
||||
Since PDUFA enactment in 1992 (PDUFA 1), the law was amended and extended in 1997 (PDUFA 2) and again in 2002 (PDUFA 3) with expiration at the end of Fiscal Year 2007. Each successive 5-year enactment has served to alter the emphasis and scope of the program, to further improve and speed new drug development and regulatory review.
As described in Section 2 of this paper, FDA funding and staffing levels for drug review activities have significantly increased under PDUFA. At the same time, the performance focus of the program has continued to expand, as summarized in the Table 4.1 below.
PDUFA 1Industry paid a fee
Goals by FY97
|
PDUFA 2Industry paid a fee
Goals by FY02
|
PDUFA 3Industry paid a fee
Goals by FY07
|
During the first few years of PDUFA 1, FDA eliminated backlogs of original applications and supplements that had formed in earlier years when the program had fewer resources. Over the course of PDUFA 1, the Agency committed to review and act on a progressively increasing proportion of original NDAs, BLAs, and efficacy supplements within 12 months and resubmissions and manufacturing supplements within 6 months. The Agency also committed to review and act on 90 percent of priority NDAs, BLAs, and efficacy supplements (i.e., submissions for products providing significant therapeutic gains) submitted in FY 1997 within 6 months. Over the course of PDUFA 1, FDA exceeded all of these performance goals.
In 1997, Congress passed the Food and Drug Administration Modernization Act of 1997 (FDAMA) and reauthorized PDUFA (PDUFA 2) for five more years. Under the PDUFA 2 program, most review times were shortened and the Agency met or exceeded nearly all its review goals. PDUFA 2 also set new goals intended to improve communication between FDA and application sponsors during the drug development process. These goals specified time frames for scheduling formal meetings with industry sponsors during the pre-clinical and clinical development phase, to review and address specific issues relating to their product. The additional goals in PDUFA 2 included commitments to respond to a sponsor's response to a clinical hold, and a commitment to review and assess special study protocols proposed by sponsors.
In 2002, Congress passed the Bioterrorism Act, which included an extension of PDUFA (PDUFA 3) for five more years, FY 2003 through FY 2007. For the first time, PDUFA 3 also authorized FDA to spend user fee funds on certain aspects of post-market risk management. The review performance and procedural goals associated with PDUFA 3 were similar to those under PDUFA 2 for fiscal year 2002 performance levels, but the PDUFA 3 program addressed drug safety issues and established several new initiatives to improve application submissions and agency-sponsor interactions during drug development and application review.
Under PDUFA 3 FDA created a guidance for review staff and industry on good review management principles and practices (GRMPs) as they apply to the first cycle review of NDAs, BLAs, and efficacy supplements. FDA announced the guidance's availability in the Federal Register of March 31, 2005 (70 FR 16507). FDA also set a goal of testing whether providing early review of selected applications and additional feedback and advice to sponsors during drug development for selected products can shorten drug development and review times.
PDUFA 3 includes two "continuous marketing application" (CMA) pilot programs; CMA Pilot 1 provides for the review of a limited number of pre-submitted portions of NDAs and BLAs. Under CMA Pilot 2, FDA and applicants can enter into agreements to engage in frequent scientific feedback and interactions during the investigational new drug phase of product development. Both the first-cycle and CMA initiatives are currently being evaluated to determine their impact on the effectiveness and efficiency of FDA-sponsor communications, product development, and regulatory review.
In addition, the goals under PDUFA 3 also included new provisions, for example, to develop guidance for industry on good risk assessment, risk management, and pharmacovigilance practices, to fund outside expert consultants to help evaluate and improve review management processes. In PDUFA 3 FDA also agreed to centralize accountability and funding for all PDUFA information technology initiatives and activities. Details of the PDUFA 3 goal commitments can be found at http://www.fda.gov/oc/pdufa/PDUFAIIIGoals.html.
FDA has seen a significant increase in drug review workload both as a result of increases in the volume of sponsor submissions for review, and due to the review performance goals specified under the PDUFA authorizing legislation.
This section summarizes trends in the workload of the PDUFA program. These include FDA review and action on:
In general, the workload is driven by the technical complexity and staffing requirements of the activities FDA must perform to address the submissions and requests; the intensity of that effort, determined by the deadlines imposed by the PDUFA performance goals; and the volume of sponsor requests and submissions.
4.2.1 Review of Original New Drug Application (NDA) and Biologics Licensing Application (BLA) Submissions
Review of NDA and BLA submissions requires an intensive multidisciplinary review, as described in Section 3.3 Under PDUFA, FDA's goal is to review and act on all filed original NDA/BLA submissions within the following time frames: within 6 months for 90 percent of all priority applications; and within 10 months for 90 percent of all standard applications.
In addition, FDA ‘s goal is to review and act on 90 percent of all NDA/BLA Class 1 resubmissions within 2 months, and 90 percent of all Class 2 resubmissions within 6 months.
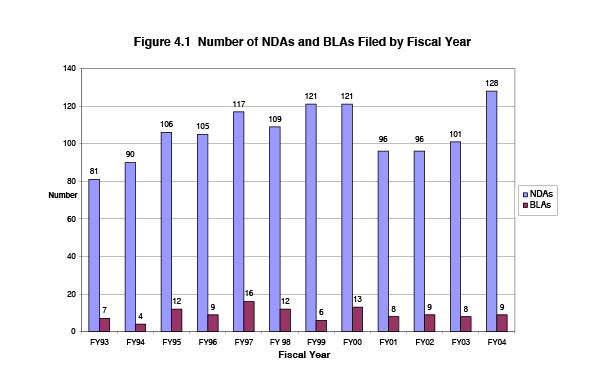
The following chart summarizes trends in the number of NDA and BLA submissions during the PDUFA era. While the number of BLA submissions has remained fairly constant during this time period, the number of NDA submissions has increased from 96 filed in FY2002, at the end of PDUFA 2, to 128 in FY 2004. (As noted earlier, about 25 percent of all applications received each year do not pay fees because the applications are for fee-exempt orphan products, or the fees are waived either because it is the first application from a small business or it qualifies for one of the other waiver provisions.)
As described earlier in Section 3.3, following approval of an original NDA or BLA, a sponsor may later submit an application to expand the disease indications included in the drug labeling. This application often includes additional clinical study data to support the proposed change in labeling, and is referred to as an Efficacy Supplement. FDA's review of Efficacy Supplements results in an action letter to the sponsor outlining FDA's decision. Under PDUFA, FDA's goal is to review and act on 90 percent of all filed original Efficacy Supplements: within 6 months for all priority efficacy supplements; and within 10 months for standard efficacy supplements.
Trends in workload for Efficacy Supplements and Manufacturing Supplements for both CDER and CBER are shown in the charts below. The number of filed Efficacy Supplements for FDA review has increased by more than 80 percent, from 100 submissions in FY 1993 to 184 submissions in FY 2004.

To obtain marketing approval, a sponsor must show not only that the product is safe and effective, but can be manufactured in large quantities while still retaining all of the qualities of the product that was tested in smaller batches during clinical trials. The details of manufacturing are described in the Chemistry and Manufacturing Controls (CMC) section of the NDA. After product approval, sponsors often continue to refine the manufacturing process. They must submit these changes for FDA review, and await FDA prior approval before implementing more significant changes proposed in CMC supplements.
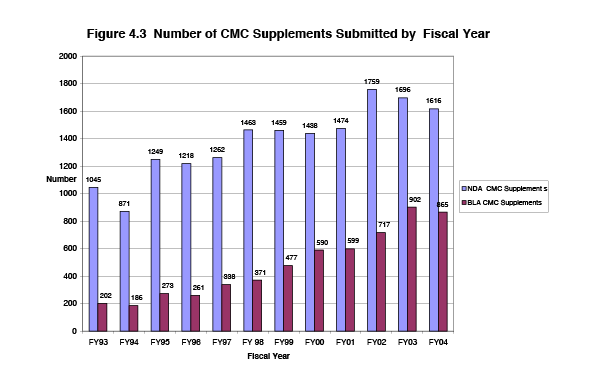
FDA's goal is to review and act on 90 percent of Changes Being Effective (CBE) CMC supplements within 6 months of receipt, and to review and act on CMC supplements requiring prior approval within 4 months of receipt. Figure 4.3 shows the trend in number of submissions of CMC supplements. Since the start of PDUFA, the number of CMC supplements has approximately doubled, from 1,247 submissions in FY 1993 to 2,481 submissions in FY 2004.
An industry sponsor can improve the quality of its drug development and related new drug application by early and more frequent consultation with FDA during drug development. For example, the sponsor can ask FDA to review its proposed clinical study designs including the starting doses for clinical trials, the type of patient population that would be enrolled and the clinical outcomes that the sponsor plans to collect during the trials, as a basis for demonstrating safety and effectiveness.
By consulting early with FDA, before investing substantial sums to conduct the clinical trials, sponsors can find out if FDA thinks their proposed design will meet (or exceed) what is necessary to demonstrate safety and effectiveness. By checking early, and revising the protocol before running the trials, sponsors can better serve participating patients and also avoid the delay and expense of conducting trials that FDA later finds are deficient and need to be augmented with additional studies.
With the increasing cost of drug development [4] and thinning pipeline of new drugs [5] more and more companies are seeking this early advice from FDA. When the sponsor requests a meeting they must also submit a background package on the specific product and application issues they wish to discuss in the meeting. Under PDUFA, FDA's goal is to schedule a formal meeting within 14 calendar days of the sponsor's request. In addition, FDA agreed to the following goals for scheduling 90 percent of requested meetings:
These formal meetings with sponsors typically involve the following tasks for FDA:
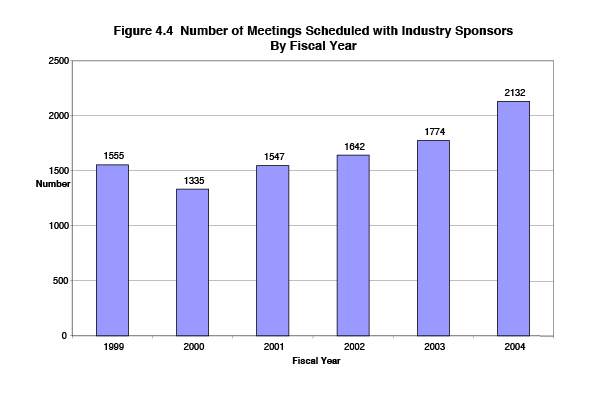
These meetings with sponsors are important to improving drug quality, but they also impose a significant additional work burden on FDA. Meetings typically require the involvement of 15 FDA staff, and the total level of effort varies from an estimated 120 to 540 staff hours, depending on the number and complexity of issues to be discussed in the meeting. Figure 4.4 shows the trend in the number of industry-requested meetings held each year since the PDUFA performance commitments were established. The number has risen from 1,555 in FY 1999 to 2,132 meetings in FY 2004. The current level of annual meetings translates into an average of 9 industry-requested meetings per business day.
Special protocol assessments provide sponsors with another valuable opportunity for early FDA feedback and advice, to improve drug development, and increase the likelihood that the submitted new drug application will include sufficient evidence of safety and effectiveness to be approved on the first 'cycle' of review. Beginning with PDUFA 2, FDA committed to evaluate certain protocols and specific issues submitted by sponsors, to assess whether the design is adequate to meet scientific and regulatory requirements identified by the sponsor.
In requesting FDA's evaluation, the sponsor must submit a limited number of specific questions about the protocol design and scientific and regulatory requirements for which they seek agreement. For example, the sponsor may submit questions about whether the dose range in the carcinogenicity study is adequate, considering the intended clinical dosage; or whether the proposed clinical endpoints are adequate to support a specific efficacy claim.
Within 45 days of receiving the protocol and specific questions, FDA will provide a written response to the sponsor that includes a succinct assessment of the protocol and answers to the questions posed by the sponsor. If FDA does not agree that the protocol design, execution plans, and data analyses are adequate to achieve the goals of the sponsor, the reasons for the disagreement will be explained in the response.
Protocols that qualify for this program include: carcinogenicity protocols, stability protocols, and Phase 3 protocols for clinical trials that will form the primary basis of an efficacy claim. In order for Phase 3 protocols to qualify for this comprehensive protocol assessment, the sponsor must have had an end of Phase 2/pre-Phase 3 meeting with the relevant FDA review division so that the division is aware of the developmental context in which the protocol is being reviewed and the questions being answered.
If a protocol is reviewed under this process and agreement with FDA is reached on design, execution, and analyses and if the results of the trial conducted under the protocol substantiate the hypothesis of the protocol, FDA agrees that the data from the protocol can be used as part of the primary basis for approval of the product. The fundamental point here is that having agreed to the design, execution, and analyses proposed in protocols reviewed under this process, FDA will not later alter its perspective on the issues of design, execution, or analyses unless public health concerns unrecognized at the time of protocol assessment under this process are evident.
Written special protocol evaluations provides extremely valuable information for sponsors, significantly reducing regulatory uncertainty and business risks in the subsequent drug development. However, the high complexity and short deadlines associated with special protocol assessment impose a significant work burden on FDA, comparable to or greater than the level of effort required to conduct sponsor-requested meetings.
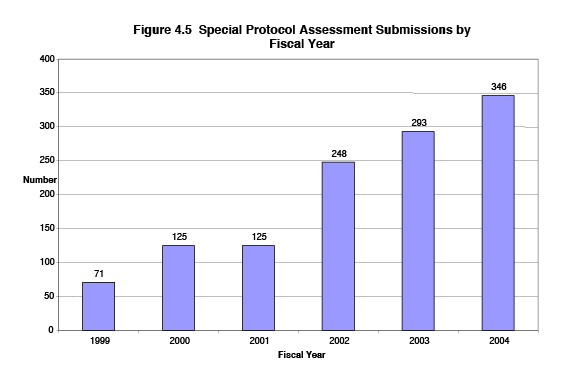
Figure 4.5 shows the trend in the number of sponsor-requested special protocol assessments. It shows the total number (CDER and CBER) of Special Protocol Assessment Receipts for each fiscal year 1999 through 2004. The number has increased by nearly 400 percent, from 71 to 346 per fiscal year during this period.
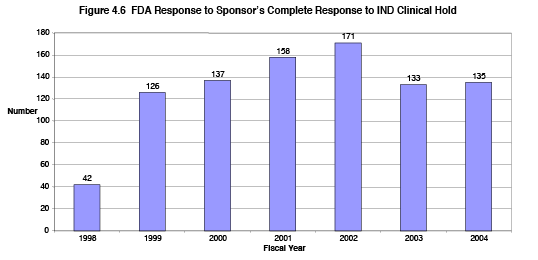
Under PDUFA, FDA's goal is to reply to a sponsor's complete response to a clinical hold within 30 days of the Agency's receipt of the sponsor's response, and do this for at least 90 percent of such submissions. As noted in earlier discussion, rapid resolution of safety issues that led to clinical hold helps ensure patient safety while enabling access to the experimental treatment. Figure 4.6 shows the total number (CDER and CBER) of Complete Response to Commercial IND Clinical Holds Received (Clinical Holds) for each fiscal year 1999 through 2004. The number of Clinical Holds increased by over 200 percent (from 42 to 135 per fiscal year) over this period.
This section describes some key resource challenges for FDA's capacity for new drug review and also for surveillance to ensure safety and effectiveness in the use of new drugs. PDUFA has been successful in making the drug review process better-staffed, more rigorous and more predictable and FDA's overall review times have been cut in half. FDA's review workload has grown very significantly, particularly during the years of PDUFA 2 and 3, with the addition of goals for sponsor-requested meetings and special protocol assessments. Starting in PDUFA 3, industry paid substantially higher fees that helped cover the costs associated with these significantly increased but highly-valued FDA activities. However, the workload levels have continued to grow, outpacing the workload adjustment of fees.
At the same time, FDA PDUFA payroll costs have far outpaced the increase in the PDUFA appropriations trigger amount and overall appropriations for drug review have been less than envisioned when PDUFA 3 was enacted. Appropriations for drug and biologics programs have actually declined since 2003, while FDA's payroll costs, the largest cost for the programs, goes up each year by 4 to 5 percent. If this trend continues FDA is uncertain about its continued ability to satisfy the PDUFA Appropriations trigger, a pre-condition for FDA's authority to collect user fees.
Over the past several years, the decline in appropriated dollars to cover the increased cost of on-board drug review "process" staff and to cover the cost of required non-payroll expenses (e.g., facilities and rent) has required that more user fee dollars be used to cover those costs. As a consequence, fewer fee dollars have been available to hire the additional staff envisioned when PDUFA 3 was enacted.
Figures 5.1 and 5.2 show overall budget trends for CBER and CDER, in nominal dollars (not adjusted for inflation) for fiscal years 2001 through 2005. Appropriated funds available to cover PDUFA costs have declined because total CDER and CBER budget authority has remained relatively flat or gone down, while payroll costs increase at 5 to 8 percent each year (as table 5.1 shows) and non-PDUFA program mandates and budget earmarks have increased. For example, CBER has received earmarks for blood safety and influenza vaccine-related activities, in addition to mandates for work in counterterrorism, tissue safety and gene therapy tracking.
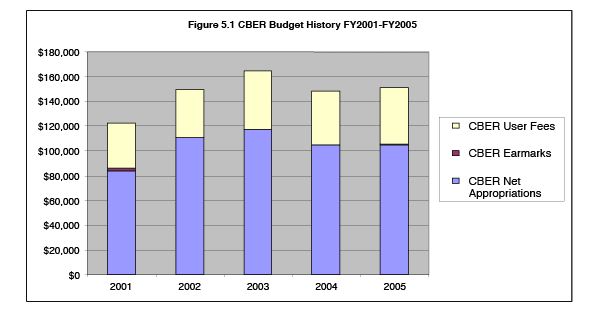
During fiscal years 2001 through 2005, CDER has receiving increased earmarks for FDA generic drug activities growing from $150,000 for generic drug education in FY 2001 to an earmark of $56 million for generic drug review in FY 2005, and mandates to increase the availability of medical countermeasures, pediatric drug labeling and address other important public health issues. The lack of growth in appropriated dollars for PDUFA has meant that appropriated dollars have not kept up with increasing payroll and non-payroll costs of the program, due to mandatory federal pay increases and the additional cost of recruiting and retaining physician disease specialists, pharmacologists, and other highly marketable senior scientific and health care professionals needed to perform regulatory review.
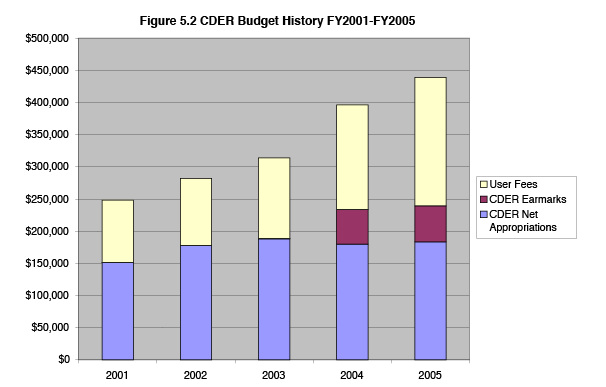
Table 5.1 below illustrates the trend in cost growth, showing payroll cost growth for CDER, the FDA product center responsible for performing approximately 90 percent of the PDUFA review work. Since 1992, the total number of center staff has grown by about 55 percent, and the average salary has more than doubled.
| Fiscal Year | Average Salary and Benefits | FTE Supported |
|---|---|---|
| FY1992 | $61,813 | 1407 |
| FY1993 | $65,287 | 1399 |
| FY1994 | $68,788 | 1473 |
| FY1995 | $71,412 | 1547 |
| FY1996 | $75,333 | 1609 |
| FY1997 | $78,371 | 1676 |
| FY1998 | $82,924 | 1647 |
| FY1999 | $87,208 | 1691 |
| FY2000 | $92,238 | 1781 |
| FY2001 | $97,710 | 1792 |
| FY2002 | $105,171 | 1788 |
| FY2003 | $111,462 | 1912 |
| FY2004 | $120,765 | 2124 |
| FY2005 | $126,700 * | 2170* |
| FY2006 | $133,000 * | 2200* |
| FY2007 | $139,700 * | 2210* |
* Figures are estimates; FY05-07 final data not yet available
At the beginning of PDUFA 3, FDA projected a constant share of appropriated funds, assuming these funds would grow at a rate that would continue to support 1277 of the FTE devoted to the review of human drug applications. But appropriations available for drug review have gone down while pay costs have gone up. Given the limited availability of appropriated funds, fee revenues have had to be used to offset cost increases at current staffing levels. The result is that FDA has been able to hire fewer additional scientific review staff to address the increasing review workload than anticipated. Figure 5.3 depicts the trend in planned versus actual FTE paid from appropriated funds versus fee collections.
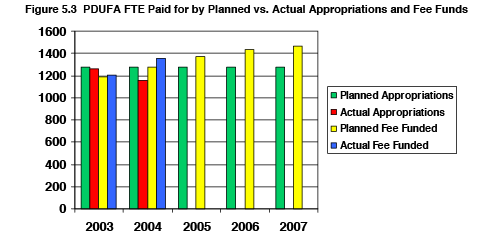
Because additional fee dollars have been applied toward the increased cost of current FTEs, fewer fee dollars have been available to hire the number of additional review staff planned under PDUFA 3, to address the underfunded workload increases of PDUFA 2, particularly related to industry-requested meetings and protocol assessments. In both of these areas workload grew further during PDUFA 3.
Although the current law provides for fee adjustments based on increases in review workload, the workload adjuster, as currently specified, fails to capture the most labor-intensive and fastest growing component of work. The current workload adjuster is based on the weighted average of the change in the total number of:
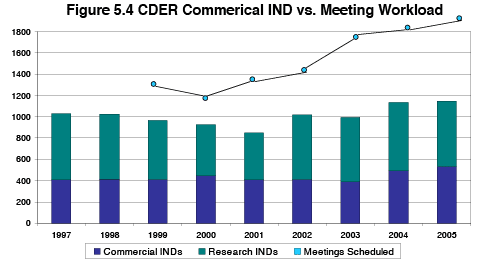
Figures 4.1 through 4.3 in Section 4 show steady but moderate growth in the 3 submission types that translate directly into PDUFA goal-driven work. By contrast, growth in the number of industry-requested meetings and special protocol assessments, which occur during the IND phase, are not correlated with, nor captured by the volume of commercial IND submissions. Figure 5.4 shows the trend in commercial INDs submittted to CDER versus the number of industry-requested meetings conducted by CDER. This graph illustrates FDA's finding that the number of submitted commercial INDs is a poor proxy for PDUFA IND-phase workload.
PDUFA 3 gave FDA new authority to allocate user fee funds to support post-marketing safety-related activities. For drugs and biologics approved on or after October 1, 2002, PDUFA 3 allows FDA to spend user fees to monitor the safety of products during their first two years on the market, three years for potentially dangerous medications.
Knowledge about a product will always be limited to some extent at the time of approval by factors in the product development process. And because of the populations not studied in clinical trials or minimally studied, side effects may be discovered if these groups are treated with a product after it goes on the market. The Adverse Event Reporting System (AERS) provides FDA's primary source of post-marketing safety data.
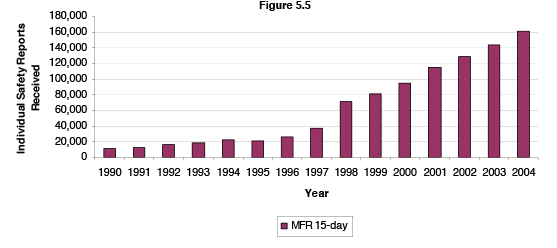
Manufacturers are required to report serious drug adverse effects to FDA within 15 days of their receipt of that report, typically from a health care provider. Figure 5.5 below shows the trend in those reports. Manufacturer 15-day reports have grown almost 9-fold from 1992 to 2004. Increases in reports may in part be the result of the increasing use of drugs, increasing patient opportunities for benefits and also increasing the exposure to risks. The number of prescriptions dispensed to US patients has grown from 1.9 billion in 1992 to 3.5 billion in calendar year 2004.[6]
FDA currently has a limited capacity to receive timely information on adverse events, and to analyze that data. For example, over 100 thousand manufacturers' 15-day reports are currently received on paper forms that must be hand-entered into FDA's electronic database. FDA needs a stronger capacity to receive timely, complete and high-quality information about patients' adverse experiences with new products, a stronger ability to quickly and accurately analyze new safety findings, and effective methods of communicating new information to patients and health care providers. These improvements require additional resources.
The budget pressures and reduced ability to spend new fees for additive capabilities, as described above, have also limited FDA's ability to significantly increase the resources available for post-market surveillance, analysis and risk management. The restriction of PDUFA funding support to products approved on or after October 2002 has also meant that fee funds could not be used to support further investigation of safety issues related to products approved within the past five to ten years, such as COX-2 and SSRI drugs that have received more recent public attention. The limited availability of fee funds and limited ability to apply them to post-market surveillance work has limited FDA's ability to address the surveillance capacity issues via PDUFA.
Industry sponsors are not required to submit Direct-to-Consumer Advertising (DTCA) materials for FDA review prior to using the materials in their promotion and this review is not part of the PDUFA "process for the review of human drugs". It is linked, however, to fundamental aspects of new drug review and it also presents the prospect of a mushrooming review workload.
A fundamental part of NDA review is the review of information that the sponsor wishes to include in the drug label, which includes the disease indication, patient population for which the drug would be approved and key safety information. FDA only approves labeling supported by scientific evidence of safety and effectiveness presented in the NDA, and any subsequent market promotion, including any direct-to-consumer advertising, must be consistent with the FDA-approved drug label and FDA regulations.
DTCA has been found beneficial to some consumers. Researchers found that of those consumers prompted by DTCA to have a physician visit to discuss treatment or seek medical advice, about 25 percent had a new diagnosis, and 43 percent of those new diagnoses were for "high priority" conditions based on federal government criteria. Some of the new diagnoses that were discovered as a result of these visits were often under diagnosed and under treated. The most common new diagnoses were allergies, high cholesterol, arthritis, hypertension, diabetes and depression[7]. Many surveyed physicians think DTCA encourages patients to seek treatments they don't need, but many also think DTCA helps educate and inform patients about disease conditions and treatments available to them[8].
Industry believes that DTCA benefits public health, and the Pharmaceutical Research and Manufacturing Association (PhRMA) has stated that DTCA "fosters an informed conversation about health, disease, and treatments between patients and their health practitioners." However, responding to some public concerns about DTCA and its potential impact on increased drug use, on August 2, 2005, PhRMA announced its Guiding Principles for Direct to Consumer Advertisements About Prescription Medicines. Principle number 8 states: "Companies should submit all new DTC television advertisements to the FDA before releasing these advertisements for broadcast."
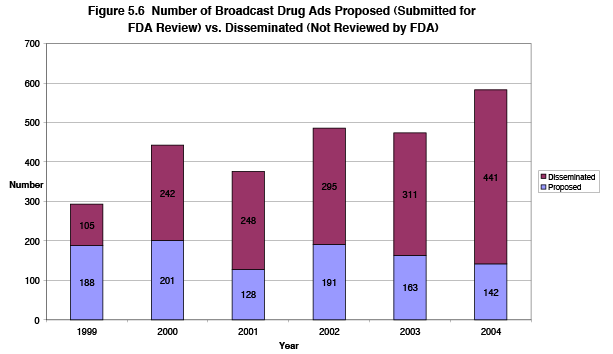
Figure 5.6 shows recent trends in the numbers of broadcast drug advertisements disseminated without FDA review, versus the number proposed and submitted to FDA for review prior to dissemination. In 2004, DDMAC reviewed 142 proposed broadcast ads, with the 4 full-time staff available to perform these reviews. Although FDA review of all materials would ensure alignment with the approved labeling and a fair balance of information on benefits and risks, current FDA resourcing for this work would probably result in delayed reviews if all companies were to submit their ads. Such delays would likely affect companies' ability to meet their marketing timelines, and discourage them from submitting the materials for prior FDA review.
[1] Discussion of the PDUFA definition of the "process of review of human drug applications" can be found at www.fda.gov/oc/pdufa/finreport2004/appendixC.html
[2] Applications for drugs similar to those already marketed are designated as "standard," while "priority" applications represent drugs offering significant advances over existing treatments.
[3] "Detailing" is a marketing practice conducted by pharmaceutical companies whereby sales representatives visit physicians' offices, explain the features of their company's drug products, and distribute advertising materials and product samples.
[4] DiMasi, Joseph A., Hansen, Ronald W., and Grabowski, Henry G., "The Price of Innovation: New Estimates of Drug Development Costs," Journal of Health Economics, 22(2), March 2003, Pages 151-185.
[5] "Does Lack of Launches Spell End of Expansion?", Scrip Magazine, February 2005, pp. 24-25
[6] Prescription Drug Trends: A Chartbook, Kaiser Family Foundation, 2000 and www.imshealth.com
[7] J.S. Weissman, D. Blumenthal, A.J. Silk, K. Zapert, M. Newman, and R. Leitman, "Consumers' Reports on the Health Effects of Direct-to-Consumer Drug Advertising", Health Affairs Web Exclusive, February 26, 2003
[8] J.S. Weissman, D. Blumenthal, A.J. Silk, M. Newman, K. Zapert, R. Leitman and S. Feibelmann, "Physicians Report on Patient Encounters Involving Direct-to-Consumer Advertising", Health Affairs Web Exclusive, April 28, 2004
![]()