|
 |
 |
|
 | |||||
| |||||||||
|
|
|
|
|
CDER Report to the Nation: 2005 Print version
2 Drug Safety and QualityIndex
HighlightsThe practical size of premarketing clinical trials means that we cannot learn everything about the safety of a medicine before we approve it. Therefore, a degree of uncertainty always exists about the risks of a medicine, not only when we approve it but also after we approve it. This uncertainty requires our continued vigilance, along with that of the industry, to collect and assess safety data for medicines on the market. As Americans are increasingly receiving the benefits of important new drugs before they are available to citizens of other countries, we must be especially vigilant in our surveillance. We also monitor the quality of marketed drugs and their promotional materials through product testing and surveillance. In addition, we develop policies, guidance and standards for drug labeling, current good manufacturing practices, clinical and good laboratory practices and industry practices to demonstrate the safety and effectiveness of drugs. Highlights of medication safety and quality activities in 2005 include: n Improving the electronic infrastructure to support real-time availability of the most up-to-date drug information for health-care providers and consumers. n Establishing and operating the Drug Safety Oversight Board. n Processing and evaluating more than 460,000 reports of adverse drug events, including more than 25,000 submitted directly from individuals. n Issuing 60 letters on violations in drug promotion and more than 600 letters to help ensure manufacturers comply with regulations concerning drug promotion. Included in the total were more than 200 letters concerning direct-to-consumer advertising. n Issuing warning and other regulatory letters to firms selling drugs that were unapproved, poorly poor manufactured or labeled incorrectly. We also supported successful federal lawsuits against a firm importing unapproved drugs and a firm distributing unapproved and poorly manufactured prescription and over-the-counter drugs.n Evaluating more than 2,800 reports concerning problems that occur in the manufacturing, processing, packing, labeling, storing or distributing drugs. n Issuing 16 Public Health Advisories regarding drug safety issues. n Approving Medication Guides for prescription non-steroidal anti-inflammatory drugs with any of 18 active ingredients and for five additional prescription drugs. Safety System for MedicinesOur current system for evaluating drug safety provides: n Extensive premarket testing with rigorous review, including evaluation of remaining uncertainties. Premarket testing cannot, however, detect very rare, serious adverse events (see below). n Risk management strategies before and after approval. n Voluntary adverse event reporting systems with additional population-based information. n User-friendly communication through an improved drug label compatible with e-prescribing and electronic decision support. Capacities of current postmarketing safety systemn Profile of common adverse events in populations studied during development. n Understanding of medication metabolism and common metabolism-based drug-drug interactions. n Management and evaluation of certain anticipated postmarketing risks. n Identification of rare serious adverse events that occur after marketing. Knowledge gaps in postmarketing safety systemn Differences in the frequency of events between those taking and not taking a drug. n Detection of events after long-term exposure. n Adverse events that occur more frequently in populations not normally studied in trials such as those who are very sick or on multiple drugs. n Adverse events that occur more frequently with off-label use. n A tendency for medical errors or abuse. Approaches to resolving knowledge gapsn Use emerging electronic medical record systems for surveillance. n Randomized trials or registries conducted in practice settings after marketing. n More surveillance systems in specialized settings such as emergency rooms or nursing homes.
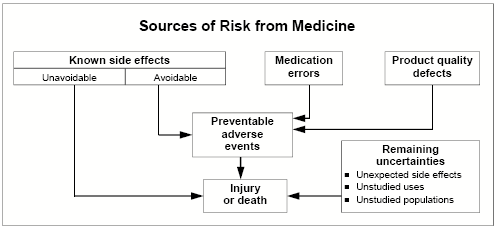
Click on image for accessible text. Types of risks from medicinesProduct quality defects. These are controlled through good manufacturing practices, monitoring and surveillance. Known side effects. Predictable adverse events are identified in the drug's labeling. These cause the majority of injuries and deaths from using medicines. Some are avoidable, and others are unavoidable. n Avoidable. Drug therapy requires an individualized treatment plan and careful monitoring. Other avoidable side effects are caused by known drug-drug interactions. n Unavoidable. Some known side effects occur with the best medical practice even when the drug is used appropriately. Examples include nausea from antibiotics or bone marrow suppression from chemotherapy. Medication errors. For example, the drug is administered incorrectly or the wrong drug or dose is administered. Remaining uncertainties. In addition to rare events occurring in about 1 in 10,000 persons,these include long-term effects and unstudied uses and populations. Drug Safety SurveillanceWe evaluate the safety of drugs available to American consumers using a variety of tools and disciplines. We maintain a system of postmarketing surveillance and risk assessment programs to identify adverse events that did not appear during the drug development process. We monitor adverse events such as adverse reactions, drug-drug interactions and medication errors. We have access to commercial databases that contain non-patient-identifiable information on the actual use of marketed prescription drugs in adults and children. This dramatically augments our ability to determine the public health significance of adverse event reports we receive. As we discover new knowledge about a drug’s safety profile, we make risk assessments and decisions about the most appropriate way to manage any new risk or new perspective on a previously known risk. Risk management methods may include new labeling, drug names, packaging, “Dear Health Care Practitioner” letters, education or special risk communications, restricted distribution programs or product marketing termination. Adverse Event Reporting SystemA powerful drug safety tool is the Adverse Event Reporting System, known as AERS. This computerized system combines the voluntary adverse drug reaction reports from MedWatch and the required reports from manufacturers. These reports often form the basis of “signals” that there may be a potential for serious, unrecognized, drug-associated events. When a signal is detected, further testing of the hypothesis is undertaken using various epidemiological and analytic databases, studies and other instruments and resources. AERS features both paper and electronic submission options, international compatibility and pharmacovigilance screening. Adverse event reportingIn 2005, we received 464,068 reports of suspected drug-related adverse events:
Click image for larger chart, click here for accessible text. Adverse event reporting complianceWe monitor the pharmaceutical industry’s submission of adverse event reports. A firm’s procedures for collection, evaluation and submission may affect the transfer and quality of safety data that we have for analysis. Our surveillance of industry is based upon the risks associated with specific drug products and specific data processing procedures. Risk-based inspectionsWe inspect drug firms' adverse event reporting based upon risk criteria associated with specific drug products and corporate performance. These include:
Inspection outcomeIn fiscal year 2005, our field investigators inspected 106 domestic and six foreign firms to assess compliance with our regulations for adverse event reporting. We sent three firms official notification that they had significant uncorrected deficiencies. We were able to work with about 40 firms to obtain voluntary correction of deficiencies identified by our monitoring. Outreach and educationIn addition to our inspectional program for adverse event compliance, we improve safety reporting through educational presentations to industry. These provide industry with a direct opportunity to expand their understanding of reporting requirements and best practices in drug safety and to alert them to pending regulatory changes. These meetings also serve to expand our own knowledge of industry’s worldwide pharmacovigilance activities. Our educational activities include formal presentations at global industry meetings and training for FDA field investigators. Adverse event electronic submissionsElectronic submission of adverse event reports permits more timely receipt and evaluation at considerable cost savings for us and industry. Our initiative to encourage electronic reports continues to make progress and remains a high priority. We provide useful information on electronic adverse event reports at http://www.fda.gov/cder/aerssub/default.htm. AERS on InternetYou can learn more about the Adverse Event Reporting System at http://www.fda.gov/cder/aers/default.htm. Data miningWe concluded work under our two-year data mining cooperative research and development agreement with a commercial firm to develop advanced software tools for quantitative analysis of drug safety data. The resulting software, WebVDME, is now in full production use by safety evaluators and epidemiologists. Data mining for simple drug-event signal generation is one potential contribution data mining and related quantitative methods can make to increase our awareness and understanding of trends and patterns in adverse drug reactions. Population-based drug safety evaluationNew contracts will help us evaluate the safety of newly marketed drugs faster and more effectively. We awarded four contracts that give us access to databases that include more than 20 million patients in different geographic areas and include special populations. The contracts, which give us more flexibility and access to a wider range of data resources than we previously had, can be used to: n Conduct safety analyses. n Respond in a timely manner to urgent public safety concerns. n Provide a mechanism for collaborative research designed to test hypotheses, particularly those arising from suspected adverse reactions reported to us. n Enable our rapid access to U.S. population-based data sources to ensure public safety when necessary. MedWatch Outreach and ReportingWe administer the MedWatch program that helps promote the safe use of drugs by: n Rapidly disseminating new safety information on the Internet and by providing e-mail notification to health professionals, institutions, the public and our MedWatch partners consisting of professional societies, health agencies and patient and consumer groups. n Providing a mechanism for health professionals and the public to voluntarily report serious adverse events, product quality problems and product use errors for all FDA-regulated medical products. Reports can be filed by mail, fax, telephone or the Internet. n Educating health professionals and consumers about the importance of recognizing and reporting serious adverse events and product problems, including medication errors. Our education program includes Internet outreach, speeches, articles and exhibits. Individual health-care professionals and consumers can subscribe to our e-mail notification service, which now has 56,000 members. We also have 160 MedWatch Partner organizations. In 2005, these individuals and groups received: n 109 safety alerts for drugs and therapeutic biologics. n 25 to 70 safety-related labeling changes for drugs each month. MedWatch Internet resourcesn You can find the latest medical product safety information at http://www.fda.gov/medwatch/. n You can sign up for immediate e-mail notification of MedWatch safety information at http://www.fda.gov/medwatch/elist.htm. Public Health Advisories in 2005We issued 16 advisories to alert health-care providers and consumers to: n Increased liver toxicity associated with the anti-HIV drug nevirapine. n Concerns about the risk of sudden death in patients treated with Adderall XR, a drug product containing amphetamines, and a Canadian action on the product. n Our warning to avoid using the epilepsy drug tiagabine in patients without epilepsy because of seizures. n The suspended marketing of natalizumab, a treatment for multiple sclerosis, which was remarketed in 2006 with a risk management plan. n Our advice that the eczema therapies pimecrolimus and tacrolimus only be used as directed—that is for short term use and not at all in children younger than 2 years of age—and only after other treatments have failed to work because of a potential cancer risk. n An increased risk for serious muscle toxicity associated with the cholesterol-lowering drug rosuvastatin, especially at the highest approved dose. n The increased risk of heart attack and stroke associated with the long-term use of non-steroidal anti-inflammatory drugs. n Increased deaths when atypical antipsychotic drugs are used to treat behavioral disorders in elderly patients. n Suicidality in adults being treated with antidepressants. n The suspended marketing of Palladone, an extended-release formulation of an opioid pain medication. n Our highlighting the importance of the safety information in the labeling when using fentanyl transdermal skin patches for pain control because of reports of death and other serious side effects. n Four cases of fatal infection in women following medical abortion with mifepristone. n Suicidal thinking in children and adolescents associated with atomoxetine, a drug approved to treat attention deficit hyperactivity disorder. n Possible increased risk of worsening wheezing and death in some people treated with several long-lasting bronchodilator medicines. n Increased risk for congenital malformations when the antidepressant paroxetine is used in the first trimester of pregnancy. n The marketing suspension of a diagnostic imaging agent. Internet resourcesLinks to our Public Health Advisories and associated information are at http://www.fda.gov/cder/news/pubpress.htm. Medication GuidesWe may require specific written patient information for selected prescription drugs that pose a serious and significant public health concern. This information is called a Medication Guide. We require Medication Guides when the information is necessary for patients to use the product safely and effectively. In 2005, we approved Medication Guides for prescription pain relievers with any of 18 nonsteroidal anti‑inflammatory ingredients (below) and these five other drugs:
These Medication Guides must be provided to patients with each prescription dispensed. NSAID MedGuide active ingredients
User fees support risk assessment, minimizationIn recent years, about half of all new medicines marketed worldwide have been launched in the United States, and American patients have had access to about three-quarters of the world’s new medicines within the first year of their introduction. The law authorizing us to collect user fees allows us to spend some of those funds to increase our assessment and minimization of risks of medicines both before they are approved and after approval: n Preapproval. Sponsors are invited to submit proposed risk management plans before they submit an application for a new drug or biologic. Our drug safety experts carefully review the proposals and begin discussions with sponsors at this early stage that continue through application review and after approval. n Postapproval. User fees also fund surveillance of the safety of medicines during their first two years on the market or three years for potentially dangerous medications. It is during this initial period, when new medicines enter into wide use, that we are most likely to identify and counter adverse side effects that did not appear during the clinical trials. Risk management guidances publishedWe published three risk management draft guidances for industry in 2005: n Premarketing Risk Assessment focuses on measures companies might consider throughout all stages of a medicine’s clinical development. n Development and Use of Risk Minimization Action Plans describes how to address specific risk-related goals and objectives and also suggests various tools to minimize the risks of drug and biological products. n Good Pharmacovigilance Practices and Pharmacoepidemiologic Assessment identifies recommended reporting and analytical practices to monitor the safety concerns and risks of medicines in general use. The three guidances fulfill our commitment to the risk management performance goals that are part of the 2002 reauthorization of the Prescription Drug User Fee Act. Medication Error PreventionAvoiding name, label, labeling and packaging confusionWe work hard to ensure the safe use of drugs we approve by weeding out brand names that look or sound like the names of existing products. We identify and avoid brand names, labels, labeling and packaging that might contribute to problems or confusion in prescribing, dispensing or administering drug products. We review about 300 post-marketing reports of medication errors each month. About half are due to error-prone labeling such as similar looking labels and labeling, poor package design, confusing instructions for use and confusing names. We investigate the causes and contributing factors of these errors and recommend revisions to the label, labeling and/or packaging of these products to avert further error. Our comprehensive Web site on
medication errors is at Bar codes required on medicinesOur regulation that calls for medicines to have a bar code became final in February 2004. It covers most prescription medicines and certain over-the-counter medicines commonly used in hospitals. The bar code rule aims to protect patients from preventable medication errors by helping ensure that health professionals give the right patient the right drug, at the appropriate dosages, at the right time. The rule will support and encourage widespread adoption of advanced information systems that, in some hospitals, have reduced medication error rates by as much as 85 percent. We estimate that the rule will help prevent nearly 500,000 adverse events and transfusion errors while saving $93 billion in health costs over 20 years. Drug ShortagesWe work to help prevent or alleviate shortages of medically necessary drug products. Drug shortages occur for a variety of reasons including manufacturing difficulties, bulk supplier problems and corporate decisions to discontinue drugs. Because drug shortages can have significant public health consequences, we work with all parties involved to make sure all medically necessary products are available within the United States. Drug shortage program aids counterterrorism effortUtilizing data obtained from manufacturers and distributors, our drug shortage program provides supply and production information in response to federal government requests in relation to counterterrorism efforts. Drug shortages on the InternetWe have a Web site that lists current drug shortages, describes efforts to resolve them and explains how to report them. n The site is at http://www.fda.gov/cder/drug/shortages. n We have an e-mail address to provide the public a communication tool for drug shortage information at DrugShortages@fda.hhs.gov.
Date created: August 18, 2006 |