 |
 |
 |
|
 |
FDA Home Page | Search
FDA Site | FDA A-Z Index | Contact
FDA | FDA Centennial
![]()
Printable version (234 KB PDF)
FY 2006

PERFORMANCE REPORT
TO CONGRESS
for the
Animal Drug User Fee Act

Food and Drug Administration
Center for Veterinary Medicine
I am pleased to present the Food and Drug Administration’s (FDA’s) FY 2006 Performance Report to Congress for the Animal Drug User Fee Act (ADUFA) of 2003. This report presents FDA’s accomplishments for FY 2006, the third year operating under ADUFA, and also updates and finalizes the FY 2005 cohort data. It is my pleasure to report that FDA met or exceeded each performance goal for FY 2005 and is meeting or exceeding each performance goal targeted for FY 2006.
FDA’s first 3 years under ADUFA have been highly productive and successful. Since FY 2004, FDA has met or exceeded all of the review performance goals established under ADUFA. This has been accomplished by such measures as hiring a substantial number of additional FDA staff, staff development activities, and the development and dissemination of guidance, policy, and procedural documents. These actions are an integral part of FDA’s commitment to improving the efficiency, quality, and predictability of the new animal drug review process to meet more demanding review time goals established under ADUFA in FY 2007 and FY 2008. In FY 2007, FDA plans to build on its accomplishments to:
FDA is committed to improving the efficiency, quality, and predictability of the new animal drug review process. We are dedicated to exploring new approaches and technologies that offer high quality and cost-effective improvements in FDA’s review of new animal drug applications and submissions. FDA looks forward to the continued success and significant improvements in the animal drug review process that ADUFA will help make achievable.
Andrew C. von Eschenbach, M.D.
Commissioner of Food and Drugs
On November 18, 2003, the President signed ADUFA into law. ADUFA amends the Federal Food, Drug, and Cosmetic Act (FD&C Act) to authorize FDA to collect user fees from new animal drug sponsors. Under ADUFA, FDA agreed to pursue a comprehensive set of review performance goals to improve the timeliness and predictability of the review of new animal drug applications (NADAs) and investigational new animal drug (INAD) submissions. This report updates and finalizes FY 2005 accomplishments and describes FDA’s accomplishments in FY 2006 toward meeting the performance goals.
FDA continues to achieve expectations in implementing ADUFA. Among the key activities and accomplishments during FY 2006 were:
FY 2006 Activities and Accomplishments
Performance At-A-Glance for FY 2005 and FY 2006
Implementation Plans for FY 2007
Report on Final FY 2005 and Preliminary FY 2006 ADUFA Cohort Performance
Supplemental NADAs and Reactivations
Abbreviated New Animal Drug Applications
Appendices:
Appendix A: HHS Secretary’s Commitment Letter to Congress
Appendix B: Summary of the ADUFA Performance Goals
Appendix C: Summary of Footnotes
ADUFA was enacted on November 18, 2003, and authorized FDA to collect user fees for certain applications and supplements, establishments, products, and sponsors to support the review of new animal drugs. The Consolidated Appropriations Act of 2004, enacted on January 23, 2004, contained an appropriations provision enabling FDA’s implementation of ADUFA. Under ADUFA, FDA agreed to meet specified performance goals for the review of certain submissions over 5 years. FDA agreed to review and act on submissions within shorter time periods for each new fiscal year. Information about ADUFA, including the text of the HHS Secretary’s November 13, 2003, commitment letter to Congress, is located in Appendix A and can also be found at: http://www.fda.gov/oc/adufa.
ADUFA requires that the Secretary submit two annual reports to Congress for each fiscal year fees are collected: 1) a performance report due within 60 days of the end of the fiscal year, and 2) a financial report within 120 days of the end of the fiscal year. This report is FDA’s third annual performance report and updates and finalizes FY 2005 cohort data. The report also summarizes FDA’s continuing progress in implementing ADUFA and in meeting quantifiable ADUFA review goals for FY 2006. This report also describes FDA’s implementation plans for FY 2007.
ADUFA was signed into law on November 18, 2003, amending the FD&C Act and providing FDA with important new responsibilities, resources, and challenges. The goal of ADUFA is to better serve animal health and public health by providing additional funds to augment FDA resources devoted to “the process for review of new animal drug applications.”
Under ADUFA, FDA agreed to meet certain review performance goals. These goals strive to expedite the review of NADAs, supplemental NADAs, and INAD submissions.
This program is similar to the Prescription Drug User Fee Act (PDUFA) program for human drugs that has been in place for 14 years. The expectation is that ADUFA, like PDUFA, will continue to help FDA expedite and improve its review of applications for new animal drugs so that safe and effective new products will be available more quickly. The guidelines and definitions below apply to FDA’s implementation of ADUFA. Further information can be found in Appendix A and can also be found at: http://www.fda.gov/oc/adufa.
Review and Act on Applications and Submissions. The term "review and act on" is understood to mean the issuance of a complete action letter after the complete review of an animal drug application, supplemental animal drug application, or investigational drug submission which either (1) approves an animal drug application or supplemental application, or notifies a sponsor that an INAD submission is complete, or (2) sets forth in detail the specific deficiencies in such animal drug application, supplemental animal drug application, or investigational animal drug submission and, where appropriate, the actions necessary to place such an application, supplemental application, or submission in condition for approval.
Refuse to File Applications and Refuse to Review Submissions. Within 30 days of submission, FDA shall “refuse to file” an animal drug application, supplemental animal drug application, or their reactivation, which is determined to be insufficient on its face or otherwise of unacceptable quality for review upon initial inspection per 21 CFR 514.110. Thus, FDA will refuse to file an application containing numbers or types of errors, or flaws in the development plan, sufficient to cause the quality of the entire submission to be questioned to the extent that FDA cannot reasonably review it. Within 60 days of submission, FDA will refuse to review an INAD which is determined to be insufficient on its face or otherwise of unacceptable quality upon initial inspection using criteria and procedures similar to those found in 21 CFR 514.110. A decision to refuse to file an application or to refuse to review a submission as described above will result in the application or submission being excluded from the cohort upon which the relevant user fee goal is based. FDA will record the numbers and types of these exclusions and include them in its annual performance report.
ADUFA established review performance goals for FDA that are being phased in over a 5 year period. These performance goals run from FY 2004 through FY 2008 and are intended to achieve progressive, yearly improvements in the review process associated with approval of new animal drug applications. FDA agreed to review and act on submissions within shorter periods of time each new year. With the fifth and final year of ADUFA ending on September 30, 2008, FDA has agreed to review and act on 90 percent of the following submission types within the specified times:
While the performance goal of reviewing 90 percent of submissions within specified times remains constant over the 5-year ADUFA period, the specified timeframes incrementally decrease over this period for all submission types. The FY 2008 review time goals will be the most challenging and difficult for FDA to meet, as review time goals decrease to the shortest number of review days for this 5-year period. The 5-year progression of these goals is presented in Appendix B.
All FDA review performance statistics are based on a fiscal year receipt cohort. This methodology calculates performance statistics for submissions for the fiscal year FDA received them, regardless of when FDA ultimately acted on or approved the submissions. A result of this approach is that the statistics shown for a particular year may change from one report to the next. This is because, as time passes, FDA completes work on more and more submissions in a receipt cohort. As more submissions are completed, the statistics for that year of receipt must be adjusted to reflect the new completions. Until all submissions in a cohort are completed, only a preliminary performance assessment can be provided for that cohort. In this report, FDA is providing the status of FY 2006 performance, as of September 30, 2006, and final performance for FY 2005.
As part of ADUFA implementation, FDA eliminated backlogs by hiring new employees, developing staff, issuing guidance to industry, and developing policy and procedure documents to improve the new animal drug review process. These actions are intended to position FDA to meet the progressively challenging performance goals of ADUFA.
FDA continued to achieve expectations in implementing ADUFA. Key activities and accomplishments during FY 2006 included:
1) Process for Developing Standard Operating Procedures (SOPs) for Office of New Animal Drug Evaluation (ONADE).
2) Refuse to File/Refuse to Review.
3) Scheduling and Holding Meetings with Outside Parties.
1) Guidance #183: Animal Drug User Fees: Fees Exceed Costs Waivers and Reductions - notice of availability of draft guidance published August 17, 2006.
2) Guidance #137: Analytical Methods Description for Type C Medicated Feeds - notice of availability of draft guidance published June 28, 2006.
3) Guidance #171: Waivers of In Vivo Demonstration of Bioequivalence of Animal Drugs in Soluble Powder Oral Dosage Form Products and Type A Medicated Articles - notice of availability of final guidance published February 17, 2006.
4) Guidance #135: Validation of Analytical Procedures for Type C Medicated Feeds - notice of availability of final guidance published November 8, 2005.
These guidance documents are available on the CVM homepage on the FDA web site at: http://www.fda.gov/cvm.
FDA met or exceeded each of the ADUFA review performance goals (90 percent on time within the specified review times) for the FY 2005 cohort.
As of September 30, 2006, FDA is meeting or exceeding each of the designated ADUFA review performance goals (90 percent on time within the specified review times) for the FY 2006 cohort acted on thus far. With submissions still pending, it is too early to make a final performance determination for FY 2006.
The table below summarizes FDA’s review performance on the FY 2005 application submissions and the preliminary performance in reviewing FY 2006 application submissions.
During FY 2006, FDA will continue to expand its efforts, through employee hiring, training, and development of guidance. These efforts will improve the timeliness and efficiency of animal drug review programs and build FDA's capacity to meet the more rigorous goals in place for future years.
This report updates FDA’s review performance for FY 2004 and presents the FY 2005 review performance for the ADUFA performance goals and commitments. All performance data are as of September 30, 2005, and calculated percentages are rounded to the nearest whole number. The following information refers to FDA performance presented in this report.
The table below summarizes the annual review time goals for original and administrative NADAs and reactivations. Over the 5-year period defined by ADUFA, the number of review days is incrementally reduced for each type of submission while the goal of reviewing 90 percent of submissions remains constant.
Submission Type |
Review Time Goal |
Performance Goal |
||||
|---|---|---|---|---|---|---|
| FY 04 | FY 05 | FY 06 | FY 07 | FY 08 | ||
Original NADAs and Reactivations |
295 days |
270 days |
230 days |
200 days |
180 days |
90% on time |
Administrative NADAs and Reactivations |
90 days |
85 days |
80 days |
70 days |
60 days |
90% on time |
FY 2006 marked a 3-year high in both the number of original (8) and administrative (13) NADAs and reactivations filed.2 Original NADAs and reactivations doubled from FY 2005 to FY 2006, returning to approximately the FY 2004 level. Administrative NADAs and reactivations increased by 44 percent from FY 2005 to FY 2006 (see graph above and table below). |
 D D |
Filings |
|||||
|---|---|---|---|---|---|
Type |
FY 04 |
FY 05 |
FY 06 |
FY 07 |
FY 08 |
Original NADAs and Reactivations |
7 |
4 |
8 |
-- |
-- |
Administrative NADAs and Reactivations |
10 |
9 |
13 |
-- |
-- |
Total |
17 |
13 |
21 |
||
FY 2005 Submissions
The 90 percent on-time ADUFA review performance goal was exceeded for all original and administrative NADAs and reactivations in FY 2005. FDA reviewed and acted on all original NADAs and reactivations within 270 days, and all administrative NADAs and reactivations within 85 days (see table below).
Submission Type |
Review Within |
Reviewed and Acted On |
Number On Time |
Percent on Time |
ADUFA Performance Goal |
|---|---|---|---|---|---|
Original NADAs and Reactivations |
270 days |
4 |
4 |
100% |
90% |
Administrative NADAs and Reactivations |
85 days |
9 |
9 |
100% |
90% |
FY 2006 Submissions
As of September 30, 2006, two of the eight original NADAs and reactivations filed in FY 2006 have been reviewed and acted on; and both had met the 230-day ADUFA review goal. Most (11 of 13) of the administrative NADAs and reactivations filed in FY 2006 had been reviewed and acted on; and all had met the 80-day ADUFA review performance goal (see table below). With submissions still pending and not overdue, it is too early to make a final performance determination for FY 2006.
Submission |
Review Within |
Reviewed and Acted On |
Number On Time |
Percent on Time |
ADUFA Performance Goal |
|---|---|---|---|---|---|
Original NADAs and Reactivations |
230 days |
2 |
2 |
100% |
90% |
Administrative NADAs and Reactivations |
85 days |
11 |
11 |
100% |
90% |
The table below summarizes the annual review time goals for non-manufacturing and manufacturing supplemental NADAs and reactivations. Over the 5-year period defined by ADUFA, the number of review days is incrementally reduced for each type of submission while the goal of reviewing 90 percent of submissions remains constant.
Submission Type |
Review Time Goal |
Performance Goal |
||||
|---|---|---|---|---|---|---|
FY 04 |
FY 05 | FY 06 | FY 07 | FY 08 | ||
Non-manufacturing Supplemental NADAs and Reactivations |
320 days |
285 days |
235 days |
200 days |
180 days |
90% on time |
Manufacturing Supplemental NADAs and Reactivations |
225 days |
190 days |
140 days |
120 days |
120 days |
90% on time |
The total number of supplemental NADAs and reactivations has increased each year from FY 2004 to FY 2006 as a result of more manufacturing supplements received. The number of manufacturing supplements increased by 35 percent from FY 2004 to FY 2006. In contrast, the number of non-manufacturing supplemental NADAs and reactivations decreased each year (see graph above and table below). |
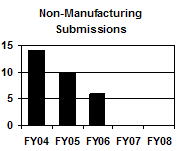 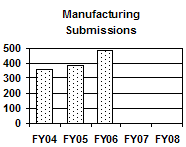 D D
|
Submissions |
|||||
|---|---|---|---|---|---|
Type |
FY 04 |
FY 05 |
FY 06 |
FY 07 |
FY 08 |
Non-manufacturing Supplemental NADAs and Reactivations |
14 |
103 |
6 |
-- |
-- |
Manufacturing Supplemental NADAs and Reactivations |
363 |
3853 |
490 |
-- |
-- |
Total |
377 |
395 |
496 |
||
FY 2005 Submissions
The 90 percent on-time ADUFA review performance goal was exceeded for all non-manufacturing and manufacturing supplemental NADAs and reactivations in FY 2005. FDA reviewed and acted on all of the non-manufacturing supplemental NADAs and reactivations within 285 days, and all but one (384 out of 385) of the manufacturing supplemental NADAs and reactivations within 190 days (see table below).
Submission Type |
Review Within |
Reviewed and Acted On |
Number On Time |
Percent on Time |
ADUFA Performance Goal |
|---|---|---|---|---|---|
Non-manufacturing Supplemental NADAs and Reactivations |
285 days |
10 |
10 |
100% |
90% |
Manufacturing Supplemental NADAs and Reactivations |
190 days |
385 |
384 |
99% |
90% |
FY 2006 Submissions
As of September 30, 2006, one of the six non-manufacturing supplemental NADAs and reactivations submitted in FY 2006 had been reviewed and acted on, and it met the 235 day ADUFA review performance goal. Nearly three-fourths (350 of 490) of the manufacturing supplemental NADAs and reactivations submitted in FY 2006 had been reviewed and acted on; and all but one (349 of 350) had met the 140-day ADUFA review performance goal (see table below). With submissions still pending and not overdue, it is too early to make a final performance determination for FY 2006.
Submission Type |
Review Within |
Reviewed and Acted On |
Number On Time |
Percent on Time4 |
ADUFA Performance Goal |
|---|---|---|---|---|---|
Non-manufacturing Supplemental NADAs and Reactivations |
235 days |
1 |
1 |
100% |
90% |
Manufacturing Supplemental NADAs and Reactivations |
140 days |
350 |
349 |
99% |
90% |
The table below summarizes the annual review time goals for INAD studies and study protocol submissions. Over the 5-year period defined by ADUFA, the number of review days is incrementally reduced for each type of submission while the goal of reviewing 90 percent of submissions remains constant.
Submission Type |
Review Time Goal |
Performance Goal |
||||
|---|---|---|---|---|---|---|
| FY 04 | FY 05 | FY 06 | FY 07 | FY 08 | ||
INAD Studies |
320 days |
285 days |
235 days |
200 days |
180 days |
90% on time |
INAD Study Protocols |
125 days |
100 days |
80 days |
60 days |
50 days |
90% on time |
The total number of these INAD submissions increased each year from FY 2004 to FY 2006. More INAD studies were submitted than INAD study protocols in FY 2004 and FY 2005, with this trend reversing in FY 2006 (see graph to the right and table below). 5 |
 D D |
Submissions |
|||||
|---|---|---|---|---|---|
Type |
FY 04 |
FY 05 |
FY 06 |
FY 07 |
FY 08 |
INAD Studies |
243 |
2955 |
221 |
-- |
-- |
INAD Study Protocols |
173 |
1506 |
262 |
-- |
-- |
Total |
416 |
445 |
483 |
||
FY 2005 Submissions
The 90 percent on-time ADUFA review performance goal was exceeded for INAD studies and study protocol submissions in FY 2005. FDA reviewed and acted on all of the INAD studies within 285 days and all but one (149 of 150) of the INAD study protocols within 100 days (see table below).
Submission Type |
Review Within |
Reviewed and Acted On |
Number On Time |
Percent on Time |
ADUFA Performance Goal |
|---|---|---|---|---|---|
INAD Studies |
285 days |
295 |
295 |
100% |
90% |
INAD Study Protocols |
100 days |
150 |
149 |
99% |
90% |
FY 2006 Submissions
As of September 30, 2006, over half (127 of 221) of the INAD studies submitted in FY 2006 had been reviewed and acted on; and all had met the 235-day ADUFA review performance goal. More than four-fifths (222 of 262) of the INAD study protocols submitted in FY 2006 had been reviewed and acted on; and all had met the 80-day ADUFA review performance goal (see table below). With submissions still pending and not overdue, it is too early to make a final performance determination for FY 2006.
Submission Type |
Review Within |
Reviewed and Acted On |
Number On Time |
Percent on Time |
ADUFA Performance Goal |
|---|---|---|---|---|---|
INAD Studies |
235 days |
127 |
127 |
100% |
90% |
INAD Study Protocols |
80 days |
222 |
222 |
100% |
90% |
Section 740(k) Abbreviated New Animal Drug Applications of the FD&C Act provides:
The Secretary shall -
“(1) to the extent practicable, segregate the review of abbreviated new animal drug applications from the process for the review of animal drug applications,” and
“(2) adopt other administrative procedures to ensure that review times of abbreviated new animal drug applications do not increase from their current level, due to activities under the user fee program.”
FDA’s CVM has established within its ONADE a separate staff, the Generic Animal Drug Team, dedicated to the review of Abbreviated New Animal Drug Applications (ANADAs) and submissions. FDA also established a team within ONADE’s Division of Manufacturing Technologies to handle related ANADA chemistry reviews.
CVM maintains a separate review queue for ANADAs. It is important to emphasize that this queue is independent from the queue maintained for the process to review NADAs under ADUFA. This ensures that ANADAs are reviewed independently of applications under ADUFA by dedicated staff. Application management processes and adherence to them are being re-examined and continue to be worked on and improved within the Generic Animal Drug Team.
To ensure that review times for ANADAs and submissions do not increase due to activities under the user fee program, ONADE established a baseline of sentinel submission review times averaged over several fiscal years (2001 through 2003). FDA staff selected document and submission types for monitoring based on submission types that were analogous to the ADUFA sentinel submission types. FDA staff continually monitors current year completed review times for these submissions. The average review times increased slightly during FY 2006. This was due, in part, to the loss of a CVM manufacturing chemistry reviewer and an additional vacancy in the Generic Animal Drug Team.
On November 13, 2003, the Department of Health and Human Services (HHS) Secretary Tommy G. Thompson sent identical performance goal letters to the following four members of Congress:
The Honorable Judd Gregg
Chairman
Committee on Health, Education, Labor and Pensions
United States Senate
The Honorable Edward Kennedy
Ranking Minority Member
Committee on Health, Education, Labor and Pensions
United States Senate
The Honorable W. J. (Billy) Tauzin
Chairman
Committee on Energy and Commerce
U.S. House of Representatives
The Honorable John Dingell
Ranking Minority Member
Committee on Energy and Commerce
U.S. House of Representatives
This appendix provides one copy of the four identical letters and a summary of the goals and procedures of CVM as agreed to under the "Animal Drug User Fee Act of 2003.".
THE SECRETARY OF HEALTH AND HUMAN SERVICES
Washington, DC, November 13, 2003
The Honorable Judd Gregg
Chairman
Committee on Health, Education, Labor and Pensions
United States Senate
Washington, DC 20510
Dear Mr. Chairman:
As you are aware, the Food and Drug Administration has been working with representatives of the veterinary pharmaceutical industry and staff of your Committee to design a new animal drug "user fee" proposal. Under this proposal, the additional revenues generated from fees paid by this industry would be dedicated for use in expediting the process for the review of animal drug applications, in accordance with performance goals that have been developed by FDA in consultation with the industry. S.313, the "Animal Drug User Fee Act of 2003" reflects the fee mechanisms developed in these discussions. The performance goals are specific in the enclosure to this letter entitled, "Animal Drug User Fee Act Performance Goals and Procedures." I believe they represent a realistic projection of what FDA can accomplish with industry cooperation and the additional resources that would be provided by the bill and annual FDA appropriations that fully cover the costs of pay and inflation increases for the animal drug review process each year.
I appreciate the support of you and your staff, and the assistance of other Members of the Committee.
Sincerely,
TOMMY G. THOMPSON
Enclosure
The goals and procedures of the FDA Center for Veterinary Medicine (CVM) as agreed to under the “Animal Drug User Fee Act of 2003” are summarized as follows:
1. Review and act on 90 percent of complete animal drug applications (NADAs) and reactivations of such applications within 180 days after submission date.
2. Review and act on 90 percent of non-manufacturing supplemental animal drug applications (i.e. supplemental animal drug applications for which safety or effectiveness data are required) and reactivations of such supplemental applications within 180 days after submission date.
3. Review and act on 90 percent of manufacturing supplemental animal drug applications and reactivations of such supplemental applications within 120 days after submissions date.
4. Review and act on 90 percent of investigational animal drug study submissions within 180 days after submission date.
5. Review and act on 90 percent of investigational animal drug submissions consisting of protocols, that the Agency and the sponsor consider to be an essential part of the basis for making the decision to approve or not approve an animal drug application or supplemental animal drug applications, without substantial data within 50 days after submission date.
Review and act on 90 percent of administrative animal drug applications (NADAs submitted after all scientific decisions have been made in the investigational animal drug process, i.e., prior to the submission of the NADA) within 60 days after the submission date.
The term "review and act on" is understood to mean the issuance of a complete action letter after the complete review of an animal drug application, supplemental animal drug application, or investigational drug submission which either (1) approves an animal drug application or supplemental application of notifies a sponsor that an investigational new animal drug submission is complete or (2) sets forth in detail the specific deficiencies in such animal drug application, supplemental animal drug application, or investigational animal drug submission in condition for approval. Within 30 days of submission, FDA shall refuse to file and animal drug application, supplemental animal drug application, or their reactivation, which is determined to be insufficient on its face or otherwise of unacceptable quality for review upon initial inspection as per 21 CFR 514.110. Thus, the agency will refuse to file an application containing numbers or types of errors, or flaws in the development plan, sufficient to cause the quality of the entire submission to be questioned to the extent that it cannot reasonably be reviewed. Within 60 days of submission, FDA will refuse to review an investigational animal drug submission which is determined to be insufficient on its face or otherwise of unacceptable quality upon initial inspection using criteria and procedures similar to those found in 21 CFR 514.110. A decision to refuse to file an application or to refuse to review a submission as describe above will result in the application or submission not being entered into the cohort upon which the relevant user fee goal is based. The agency will keep a record of the numbers and types of such refusals and include them in its annual performance report.
FDA may request minor amendments to animals drug applications, supplemental animal drug applications, and investigational animal drug submissions. At its discretion, the Agency may extend an internal due date (but not a user fee goal) to allow for the complete review of an application or submission for which a minor amendment is requested. If a pending application is amended with significant changes, the amended application may be considered resubmitted, thereby effectively resetting the clock to the date FDA received the amendment. The Agency intends to establish the same policy for investigational animal drug submissions.
Sponsors are not required to submit study protocols for review. However, for each voluntarily submitted protocol for a study that the Agency and the sponsor considered to be an essential part of the basis for making the decision to approve or not approve an animal drug application or supplemental animal drug application, the Agency will issue an acknowledgment letter providing comments resulting from a complete review of the protocol. The acknowledgement letter will be as detailed as possible considering the quality and level of detail of the protocol submission; will include a succinct assessment of the protocol; and will state whether the Agency agrees, disagrees, or lacks sufficient information to reach a decision that the protocol design, execution plans and data analyses are adequate to achieve the objectives of the study. If the Agency determines that a protocol is acceptable, this represents an agreement that the data generated by the protocol can be used to support a safety or effectiveness decision regarding the subject animal drug. The fundamental agreement is that having agreed to the design, execution, or analyses proposed in protocols reviewed under this process, the Agency will not later alter its perspectives on the issues of design, execution or analyses unless public or animal health concerns unrecognized at the time of protocol assessment under this process are evident.
1. Review and act on pending animal drug applications, supplemental animal drug applications, and investigational animal drug submissions within 24 months of initiation of user fee payments.
1. Fifty percent of FDA incremental review staff recruited and on-board by first quarter of FY 2006. Total staff increment on-board by end of FY 2008.
2. FDA will review all submissions in accordance with procedures for working within a queue. An Application/submission that is not reviewed within the applicable interim Application/Submission Goal time frame (noted below) will be reviewed with the highest possible priority among those pending.
FY 04 Review and Act on 90 percent of:
FY 05 Review and Act on 90 percent of:
FY 06 Review and Act on 90 percent of:
FY 07 Review and Act on 90 percent of:
FY 08 Review and Act on 90 percent of:
Activity |
Performance Level |
FDA Review Time |
||||
|---|---|---|---|---|---|---|
FY 04 |
FY 05 |
FY 06 |
FY 07 |
FY 08 |
||
Application/Submission Goals |
||||||
Animal drug applications (NADAs) and reactivations of such applications |
90% |
295 |
270 |
230 |
200 |
180 |
Non-manufacturing supplemental animal drug applications and reactivations of such supplemental applications |
90% |
320 |
285 |
235 |
200 |
180 |
Manufacturing supplemental animal drug applications and reactivation of such supplemental applications |
90% |
225 |
190 |
140 |
120 |
120 |
Investigational animal drug study submissions |
90% |
320 |
285 |
235 |
200 |
180 |
Investigational animal drug submissions consisting of protocols, that the agency and the sponsor consider to be an essential part of the basis for making the decision to approve or not approve an animal drug application or supplemental animal drug application, without substantial data |
90% |
125 |
100 |
80 |
60 |
50 |
Administrative animal drug applications (administrative NADAs) |
90% |
90 |
85 |
80 |
70 |
60 |
Interim Backlog Goals |
||||||
Review and act on pending animal drug applications, supplemental animal drug applications, and investigational animal drug submissions within 24 months of initiation of user fee payments. |
||||||
Additional Interim Goals |
||||||
Fifty percent of FDA incremental review staff recruited and on-board by first quarter of FY 2006. Total staff increment on-board by end of FY 2008. |
||||||
FDA will review all submissions in accordance with procedures for working within a queue. An application/submission that is not reviewed within the applicable Interim Application/Submission Goal timeframe will be reviewed with the highest possible priority among those pending. |
||||||
1. Over the 5-year period defined by ADUFA, the number of review days is incrementally reduced for each type of submission. The 5-year progression of these goals is presented in Appendix B.
2. The count of FY 2006 submissions assumes that all submissions received in the last month of FY 2006 are filed. FDA makes a filing decision within 30 days of receiving an original application. FDA calculates ADUFA review times, however, from the original receipt of the filed application.
3. One non-manufacturing supplement and one manufacturing supplement, originally reported in the FY 2005 ADUFA Performance Report, were withdrawn by the sponsor.
4. Calculated percentages are rounded to the nearest whole number up to 99 percent. Percentages above 99 percent, but below 100 percent, are rounded down to 99 percent.
5. There were 296 INAD studies that were originally reported as received in the FY 2005 ADUFA Performance Report. The final number actually received was 302. However, three studies were filed without reply; three were refused to review; and one review was stopped at the request of the sponsor. Therefore, seven studies were removed from the FY 2005 cohort.
6. In FY 2005, 169 protocols were reported as received. However, one submission was recoded as a protocol and added to the FY 2005 cohort. Thirteen protocols were filed without reply; three were refused to review; and four reviews were stopped at the request of the sponsor and, therefore, removed from the FY 2005 cohort.
 Department of Health and Human Services
Department of Health and Human Services 
Food and Drug Administration
This report was prepared by FDA's Center for Veterinary Medicine (CVM) in collaboration with the Office of Planning. For information on obtaining additional copies contact: Office of Planning (HFP-10) This report is available on the FDA Home Page at http://www.fda.gov. |
Edited September 4, 2007
![]()
